by D.A. Geier [a], P.G. King [ b] & M.R. Geier [c] [1]
Copyright © 2009 The Author(s). Published by Taylor & Francis
Published online: 11 Jun 2009.
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.
To cite this article: D.A. Geier , P.G. King & M.R. Geier (2009) Mitochondrial dysfunction, impaired oxidative-reduction activity, degeneration, and death in human neuronal and fetal cells induced by low-level exposure to thimerosal and other metal compounds, Toxicological & Environmental Chemistry, 91:4, 735-749, DOI: 10.1080/02772240802246458
To link to this article: http://dx.doi.org/10.1080/02772240802246458
Mitochondrial dysfunction, impaired oxidative-reduction activity, degeneration, and death in human neuronal and fetal cells induced by low-level exposure to thimerosal and other metal compounds
by D.A. Geiera, P.G. Kingb and M.R. Geierc [1]
(Final version received 4 June 2008)
Thimerosal (ethylmercurithiosalicylic acid), an ethylmercury (EtHg)-releasing compound (49.55% mercury (Hg)), was used in a range of medical products for more than 70 years. Of particular recent concern, routine administering of Thimerosal-containing biologics/childhood vaccines have become significant sources of Hg exposure for some fetuses/infants. This study was undertaken to investigate cellular damage among in vitro human neuronal (SH-SY-5Y neuroblastoma and 1321N1 astrocytoma) and fetal (nontransformed) model systems using cell vitality assays and microscope-based digital image capture techniques to assess potential damage induced by Thimerosal and other metal compounds (aluminum (Al) sulfate, lead (Pb)(II) acetate, methylmercury (MeHg) hydroxide, and mercury (Hg)(II) chloride) where the cation was reported to exert adverse effects on developing cells. Thimerosal-associated cellular damage was also evaluated for similarity to pathophysiological findings observed in patients diagnosed with autistic disorders (ADs). Thimerosal-induced cellular damage as evidenced by concentration- and time-dependent mitochondrial damage, reduced oxidative–reduction activity, cellular degeneration, and cell death in the in vitro human neuronal and fetal model systems studied. Thimerosal at low nanomolar (nM) concentrations induced significant cellular toxicity in human neuronal and fetal cells. Thimerosal-induced cytoxicity is similar to that observed in AD pathophysiologic studies. Thimerosal was found to be significantly more toxic than the other metal compounds examined. Future studies need to be conducted to evaluate additional mechanisms underlying Thimerosal-induced cellular damage and assess potential co-exposures to other compounds that may increase or decrease Thimerosal-mediated toxicity.
Keywords: autism; glial; lead; mercury; mercuric; neurodevelopmental
Introduction
Thimerosal (ethylmercurithiosalicylic acid) is an ethylmercury (EtHg)-releasing compound that has been used in a range of medical products for more than 70 years (Geier et al. 2007). Thimerosal contains 49.55% mercury (Hg) and, in aqueous solutions, is known to breakdown into EtHg hydroxide and the sodium salt of thiosalicylic acid (Tan and Parkin 2000).
Of particular recent concern, research showed that routine administration of Thimerosal-containing childhood vaccines resulted in some infants receiving 450% of their cumulative Hg exposure from these vaccines given during the first six months of life (Bigham and Copes 2005). In addition, these researchers found that the cumulative Hg doses (combined environmental and medicinal sources) some infants received resulted in average daily Hg doses per kilogram bodyweight that were in excess of the established US Environmental Protection Agency (EPA), Health Canada, World Health Organization (WHO), Agency for Toxic Substances and Disease Registry (ATSDR), and US Food and Drug Administration (FDA) daily safety limits for ingested Hg, with several exposures to large bolus doses of Hg resulting from Thimerosal-containing vaccine administration, during the first six months of life.
Previous studies reported that Hg exposure in any form may produce immune, sensory, neurological, motor, and behavioral dysfunctions similar to traits defining or associated with autistic disorders (ADs), and that these similarities extend to neuroanatomy, neurotransmitters, and biochemistry (Environmental Working Group 2004; Kern and Jones 2006; Mutter et al. 2005; 2007; Zahir et al. 2005). In addition, researchers from the US National Institute of Environmental Health Sciences (1999) and Nelson (1991) from the National Institute for Occupation Safety and Health of the Centers for Disease Control and Prevention described a role for Hg exposure in the pathogenesis of autism. The Collaborative on Health and the Environment’s Learning and Developmental Disabilities (2008) recently published a consensus statement reporting that there is no doubt that Hg exposure may produce autism spectrum disorders. Presumptively, Hg poisoning has also been diagnosed as autism of unknown etiology in some cases until Hg poisoning was established (Chrysochoou et al. 2003), and other researchers identified significant increases in Hg toxicity biomarkers in patients diagnosed with ADs (Geier and Geier 2006b, 2007b).
A series of recent epidemiological studies found a significant relationship between Hg exposure from routinely administered Thimerosal-containing drugs (vaccines and Rho(D)-immune globulin preparations) and an increasing risk for autism and other neurodevelopmental delays (Geier and Geier 2006a, 2006c, 2007c; Geier et al. 2008; Marques et al., in press; Young et al., in press). It was also reported in clinical studies that the greater and the earlier a child’s cumulative exposure to Hg from Thimerosalcontaining vaccines and biologics, the more clinically severe the AD (Geier and Geier 2007a). In addition, epidemiological studies of environmental toxic exposures revealed that Hg exposure was a significant dose-dependent risk factor for a child being diagnosed with an AD (Counter et al. 2002; Palmer et al. 2006, in press; Rury 2006; Windham et al. 2006).
The purpose of the present study was to evaluate the potential for Thimerosal-induced cellular damage among in vitro human neuronal and fetal model systems using cell vitality assays and microscope-based digital image capture and evaluation techniques. Furthermore, the purpose of the present study was to evaluate potential Thimerosalassociated cellular damage in the context of pathological findings observed in patients diagnosed with autistic disorders, as well as to compare Thimerosalassociated cellular damage with other toxic metal compounds (aluminum (Al) sulfate, lead (Pb) acetate, methylmercury (MeHg) hydroxide, and Hg(II) chloride) that were reported to be neurodevelopmental toxins by other investigators (Toimela and Tahti 2004; Waly et al. 2004).
Materials and methods
Human cell cultures
Cultures of SH-SY-5Y human neuroblastoma and 1321N1 human astrocytoma cells from the European Collection of Cell Cultures were purchased from Sigma-Aldrich (St. Louis, MO, USA). Nontransformed human fetal cell cultures were established from a single anonymous amniotic fluid cell sample. This sample was obtained from amniocentesis discarded waste. Upon culturing, visual inspection under an inverted UNITRON microscope (Bohemia, NY, USA) indicated that the cell type was most probably developing human epithelial cells.
For neuroblastoma cells, the culture medium consisted of Dulbecco’s Modified Eagle’s Medium/Ham’s F12, 50/50 1X with L-glutamine (MEM-F12) (Mediatech, Inc., Manassas, VA, USA), 15% fetal bovine serum (FBS) sterile filtered (Equitech-Bio, Inc., Kerrville, TX, USA), and 1% MEM nonessential amino acid (MEM NEAA) solution 100X (Sigma- Aldrich). For astrocytoma cells, the culture medium consisted of Minimum Essential Medium, Alpha 1X with Earle’s salts, ribonucleosides, deoxyribonucleosides, and L-glutamine (MEM-a ) (Mediatech, Inc.), 10% FBS, and 1% MEM NEAA solution 100X. For the fetal cells, the culture medium consisted of CHANG MEDIUM® D with L-glutamine (Irvine Scientific, Santa Ana, CA, USA) and 20% FBS. In all cases, the cells were grown following a standardized procedure at 37 C, 95% humidity, and 5% CO2 in 40mL tissue culture (NunclonTM delta surface) flasks (NUNC™, Rochester, NY, USA). Cells were grown in flasks until nearly confluent and then were trypsinized (Trypsin, INTERGEN Company, Purchase, NY, USA). The disaggregated cells were seeded evenly into COSTAR (Corning International, Corning, NY, USA) 96-well [100 uL well -1], cell-culture- cluster, flat-bottom, tissue-culture, treated plates with lid. Prior to treatment with the compounds under study, the cell aliquots seeded in each well were grown following a standardized procedure for at least one day at 37 C, 95% humidity, and 5% CO2 in the 96- well cell culture plates with appropriate cell media and FBS concentration.
Compounds
Thimerosal (C9H9HgO2SNa, CAS No. 54-64-8), aluminum sulfate octadecahydrate (Al2(SO4)3 18H2O, CAS No. 7784-31-8), Hg(II) chloride (HgCl2, CAS No. 7487-94-7), and Pb (II) acetate trihydrate (Pb(C2H3O2)2, CAS No. 6080-56-4) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MeHg(II) hydroxide, 1M in H2O (CH3HgOH, CAS No. 1184-57-2) was purchased from Alfa Aesar , a Johnson Matthey Company (Ward Hill, MA, USA). The compounds tested in the present study were highly purified, and were presumed to be > 95% pure. They were chosen for maximum solubility in aqueous environments and resistant to destabilization by oxidation or reaction with other gases that dissolve in water (CO2).
Stock solutions were prepared for each compound by dissolving them into or appropriately diluting them with MEM-a culture medium, and the resultant solutions were sterilized by filtration through a pre-sterilized 0.20 um NALGENE® Filter Unit (Nalge Nunc International, Rochester, NY, USA). The stock solutions prepared and utilized in the present study were freshly prepared for each compound tested.
Determination of cytotoxicity
Thimerosal-induced mitochondrial dysfunction in different cell types
Mitochondrial dysfunction in human neuroblastoma, astrocytoma, and fetal cells were assessed using the colorimetric 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium- 5-carboxyanilide inner salt (XTT) cell assay kit (TOX-2, Sigma-Aldrich). After at least 24 h of growth, the original media was removed from each well in the 96-well cellculture- plates, and replaced with 200 uLwell 1 dilutions of Thimerosal (10 nM–10 uM) in appropriate cell media. For the control wells (containing no Thimerosal), the same procedure was followed except 200 uL of the appropriate cell media without adding Thimerosal. The resultant 96-well cell-culture plates were covered and incubated following a standardized procedure for 24 h at 37° C, 95% humidity, and 5% CO2. The media was then removed from each well, and 50 uL of XTT solution (20% concentration, dissolved in appropriate cell media) were added to each well. The 96- well cell-culture plates were transferred to a VERSAmax tunable microplate reader (Molecular Devices, Sunnyvale, CA, USA) for assaying. The 96-well cell-culture plates were maintained at 37 ° C and were shaken for 10 s every 15 min. The contents of the study wells in the 96-well culture plates were assayed at 2 h for absorption at 450 and 690nm using SoftMax® Pro 5 software (Molecular Devices, Sunnyvale, CA, USA). The test dilutions and the controls were each repeated in eight wells. The 690nm absorbance value was subtracted from the 450nm absorbance value to determine a measure of the level of mitochondrial function of the cells in each well evaluated. The net values determined for each Thimerosal dilution were normalized to the average value for the controls, which was set at 100%. The mean results and their uncertainties (standard error of mean [SEM]) were expressed in terms of percentage control mean
([meanTest +-SEMTest] /meanControl x 100%).
Mitochondrial dysfunction in human neuroblastoma cells induced by other compounds studied
Mitochondrial dysfunction in human neuroblastoma was assessed using the XTT cell survival assay kit. The original media was removed from each well in the 96-well cellculture plates, and was replaced with 200 uLwell-1 dilutions of the following compounds: Thimerosal (10 nM–10 uM), MeHg hydroxide (100 nM–10 uM), Hg chloride (10–100 uM), Pb acetate (10–100 uM), and Al sulfate (100 uM–10 uM), each metal was dissolved in the appropriate cell media. In the control wells (containing no added compound), the same procedure was followed except no dilution of the compound being studied was added. Cells were incubated following a standardized procedure for 24 h at 37 C, 95% humidity, and 5% CO2 in the 96-well cell culture plates. The media was then removed from each well, and 50 uL of XTT solution (20% concentration, dissolved in appropriate cell media) were added to each well. The 96-well cell-culture plates were transferred to a VERSAmax for assaying. The 96-well cell-culture plates were maintained at 37° C and were shaken for 10 s every 15 min. The contents of the study wells in the 96- well culture plates were assayed at 2 h for absorption at 450 and 690nm using SoftMax® Pro 5 software. The test dilutions and the controls were each repeated in eight wells. The 690nm absorbance value was subtracted from the 450nm absorbance value to determine a measure of the level of mitochondrial function of the cells in each well evaluated. The net values determined for each dilution of the compound being examined were normalized to the average value for the controls, which was set at 100%. The results were expressed in terms of percentage control mean
([meanTest +-SEMTest]/ meanControl 100%).
Thimerosal-induced oxidative–reduction damage in human neuroblastoma cells
Oxidative–reduction damage was assessed in human neuroblastoma cells using the colorimetric resazurin-based cell survival assay kit (TOX-8, Sigma-Aldrich). The original media was removed from each well in the 96-well cell-culture plates and was replaced with 200 uLwell-1 dilutions of Thimerosal (1 nM–10 uM) in appropriate cell media. In the control wells, the same procedure was followed, except that the media added contained no added Thimerosal. Cells were incubated following a standardized procedure for 0 and 48 h at 37° C, 95% humidity, and 5% CO2 in the 96-well cell-culture plates. Subsequently, 50 mL of resazurin-based solution (10% concentration, dissolved in appropriate cell media) was added to each well. The 96-well cell-culture plates were shaken for 10 s, and incubated for 12 h at 37° C, 95% humidity, and 5% CO2.
The 96-well cell-culture plates were transferred to a VERSAmax (maintained at 37° C) for assaying. The contents of the study wells in the 96-well culture plates were then assayed for absorption at 600 and 690 nm. The test dilutions and the controls were each repeated in eight wells. The 690nm absorbance value was subtracted from the 600nm absorbance value to determine a measure of the level of oxidative–reduction activity in each well evaluated. The net values determined for each Thimerosal dilution were normalized to controls set at 100%. From the 0 and 48 h incubation times, the results (mean and SEM) were expressed as percentage control mean
([meanTest +- SEMTest]/meanControl 100%).
Visual degeneration/death induced in human neuroblastoma and human fetal cells
Cellular degeneration in human neuroblastoma and human fetal cells were assayed using microscope inspection with microscope digital image capture. The original media was removed from each well in the 96-well cell-culture plates. The media was replaced with 200 mLwell 1 dilutions of Thimerosal that were added at about the LC50 derived from the mitochondrial dysfunction assay (10–100 nM) in appropriate cell media. Further, in another series of experiments on human neuroblastoma cells, the media was replaced with 200 mLwell 1 dilution of MeHg hydroxide that was added at about the LC50, derived from the mitochondrial dysfunction assay (1–10 mM) in appropriate cell media. In control wells (containing no added test compound), the same procedure was followed except only the appropriate cell media was added. Cells were incubated following a standardized procedure for 24 h at 37° C, 95% humidity, and 5% CO2 in the 96-well cell-culture plates. The wells were then assayed using an inverted UNITRON® microscope with the 5x objective. A Big Catch™ digital eye-piece camera (EM-035M) and Scope Photo Image software (Torrance, CA, USA) were used to capture digital images of the cells.
Statistics
The statistical packages contained in StatsDirect Version 2.4.2 (Cheshire, UK) and SigmaPlot Version 9.0 (San Jose, CA, USA) were used in the present study. Dunnett’s oneway analysis of variance (ANOVA) for multiple comparisons with a control test statistic was used, and a p-value < 0.05 was considered statistically significant.
Additionally, LC50s were determined for the mitochondrial dysfunction and oxidative– reduction activity assays for the different cell types and compounds tested in the present study. The linear regression test statistic from StatsDirect was utilized to examine the linear portion of the curves derived from the assay response curves developed for mitochondrial dysfunction and oxidative–reduction activity for the different cell types and compounds tested to determine each LC50.
Results
Figure 1 presents the mitochondrial dysfunction measured at 24 h following exposure of human neuroblastoma, astrocytoma, or fetal cells to increasing Thimerosal concentrations. Thimerosal at the concentrations employed (10 nM–10 uM) in each cell type examined resulted in a significant increase in mitochondrial dysfunction relative to unexposed controls. In addition, the observed order of the sensitivity of the cell types to Thimerosal-induced mitochondrial dysfunction was human fetal cells4human neuroblastoma cells4human astrocytoma cells.
Figure 2 depicts the mitochondrial dysfunction measured at 24 h following exposure of human neuroblastoma cells to Thimerosal, MeHg hydroxide, Hg chloride, Pb acetate, and Al sulfate. The LC50 levels were determined to be as follows: Thimerosal LC50 = 82.2 nM, MeHg hydroxide LC50 = 5.6 uM, Hg chloride LC50 = 59.5 uM, Pb acetate LC50 >100 uM, and Al sulfate LC50>10mM. The observed relative order of the toxicity of the compounds to the neuroblastoma cells was Thimerosal >MeHg hydroxide > Hgchloride > Pb acetate > Al sulfate.
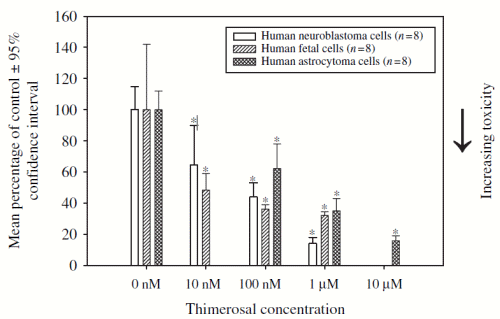
Figure 1. A concentration-dependent assessment of Thimerosal induced mitochondrial dysfunction in human cells lines following 24 h incubation. Notes: Mitochondrial dysfunction was measured using the XTT cell assay (following 2 h incubation). *p<0.05 (Thimerosal exposure concentration in comparison with the 0 nM control). Human neuroblastoma cells LC50=82.2 nM, human fetal cells LC50=9.7 nM, human astrocytoma cells LC50=337 nM.
Figure 3 presents the concentration- and time-dependent assessment of cellular oxidative–reduction activity in human neuroblastoma cells following Thimerosal exposure. Overall, Thimerosal-induced concentration- and time-dependent reduced cellular oxidative–reduction activity in human neuroblastoma cells. Specifically, the immediate exposure of human neuroblastoma cells to Thimerosal resulted in a calculated LC50=10.3 mM for cellular oxidative–reduction activity in human neuroblastoma cells. In contrast, following a 48 h incubation with Thimerosal, the observed LC50=7.6nM for cellular oxidative–reduction activity in human neuroblastoma cells.
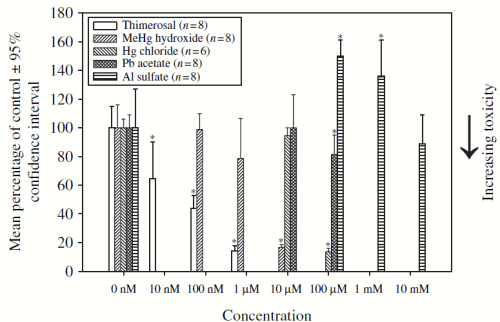
Figure 2. A concentration-dependent assessment of metal-induced mitochondrial dysfunction in human neuroblastoma cells following 24 h incubation. Notes: Mitochondrial dysfunction was measured using the XTT cell assay (following 2 h incubation). *p<0.05 (Exposure concentration in comparison with the 0 nM Control). Thimerosal LC50=82.2 nM, MeHg hydroxide LC50=5.6 mM, Hg chloride LC50=59.5 uM, Pb acetate LC50>100 uM, Al sulfate LC50>10mM.
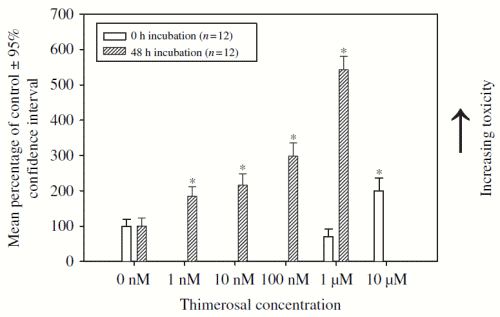
Figure 3. A concentration- and time-dependent assessment of Thimerosal induced cellular oxidative-reduction activity in human neuroblastoma cells. Notes: Cellular oxidative–reduction activity was measured using the Resazurin Dye Cell Viability Assay (following 12 h incubation). *p<0.05 (Thimerosal exposure concentration in comparison with the 0 nM Control). Thimerosal LC50=10.3 uM (following 0 h incubation) and 7.6nM (following 48 h incubation).
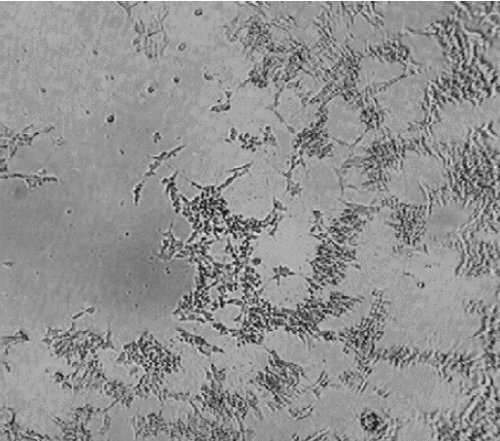
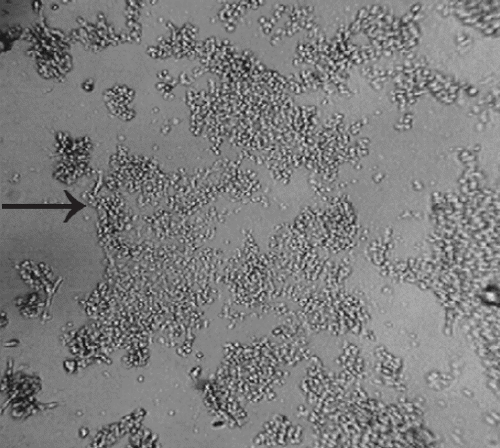
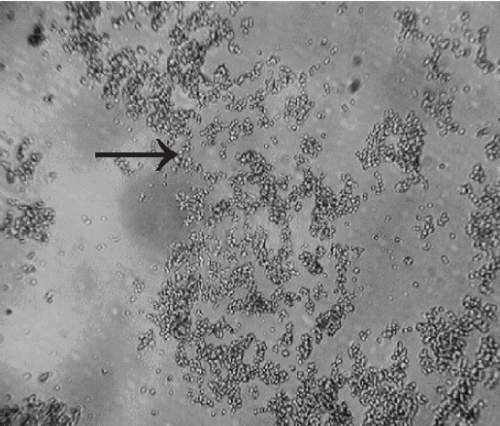
Figure 4. Images of human neuroblastoma cells following 24 h incubation. a) Neuroblastoma cells exposed to 0 nM Thimerosal. b) Neuroblastoma cells exposed to 100nM Thimerosal. c) Neuroblastoma cells exposed to 1 uM MeHg hydroxide. → – indicates cells that have undergone cellular degeneration and cellular blebbing.

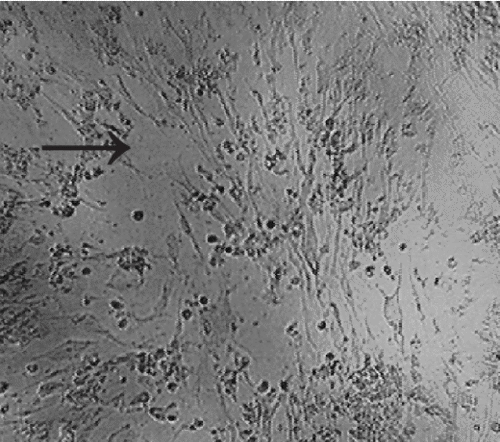
Figure 5. A visual evaluation of human fetal cells following 24 h incubation with various treatments. a) Human fetal cells exposed to 0 nM Thimerosal. b) Human fetal cells exposed to 10nM Thimerosal. → – indicates cells that have undergone cellular degeneration, cellular blebbing, and cell death.
Figure 4 shows microscope digital image captures following the application of 10–100nM Thimerosal or 1–10 mM MeHg hydroxide to human neuroblastoma cells for 24 h in comparison to unexposed controls. Visually, the addition of Thimerosal and MeHg hydroxide at concentrations consistent with the LC50 for mitochondrial-induced dysfunction in human neuroblastoma cells was associated with marked human neuroblastoma cellular degeneration in comparison with unexposed controls. Furthermore, Figure 5 shows microscope digital image captures following the 10–100nM Thimerosal to human fetal cells for 24 h in comparison to unexposed controls. Visually, Thimerosal at concentrations consistent with the LC50 for mitochondrial-induced dysfunction in human fetal cells was associated with marked human fetal cell death/inhibition of cell growth in comparison with unexposed controls.
Discussion
Cellular toxicity
In considering the potential for an exposure to induce a disorder, it is important to demonstrate that the exposure can induce the pathological findings, characterizing the disorder in an in vitro model system. Furthermore, estimating the amount of exposure necessary to induce the pathology observed in the disorder, and evaluating this in the context of other exposures that may produce and/or contribute to the disorder is essential.
The present study was principally designed to evaluate the potential for Thimerosal-induced cellular damage among in vitro human neuronal and fetal model systems using cell vitality assays and microscope-based digital image capture techniques in the context of the pathological findings observed in ADs. The results of the present study showed that Thimerosal was able to induce significant mitochondrial dysfunction, reduced cellular oxidative–reduction activity, cell death, and cellular degeneration in a concentration- and time-dependent fashion. The LC50 for mitochondrial dysfunction following 24 h incubation with Thimerosal ranged between 9.7 and 337 nM. Additionally, following 48 h of incubation with Thimerosal containing media, the LC50 for cell oxidative–reduction activity in the neuroblastoma cells studied was 7.6 nM Thimerosal. Furthermore, the different cell types showed different levels of susceptibility to Thimerosal-induced cellular toxicity. The observed order of the sensitivity of the cell types studied to Thimerosal-induced cellular toxicity was human fetal cells >human neuroblastoma cells>human astrocytoma cells.
The concentrations of Thimerosal, which induced cellular toxicity in the present study, are quantitatively consistent with previous in vitro cell model systems. Parran et al. (2005) described the LC50 for cell death in SH-SY-5Y human neuroblastoma following incubation with Thimerosal (without serum) were 38.7 and 4.35nM at 24 and 48 h, respectively. Similarly, Yel et al. (2005) demonstrated that Thimerosal, in a concentrationand time-dependent manner, even in nanomolar concentrations as low as 25nM (these researchers did not examine any concentrations of Thimerosal lower than 25 nM), significantly increased cell death in neuroblastoma cells. These researchers demonstrated that the cell death induced by Thimerosal was visually characterized by similar visual phenomena as those observed in the present study, including nuclear morphology of apoptosis, vacuolization, and chromatin condensation and shrinking. Further, Humphrey et al. (2005) reported on mitochondrial mediated Thimerosal-induced apoptosis in SKN- SH human neuroblastoma cells. These researchers visually observed that low-concentration Thimerosal exposure rapidly induced neuronal cell degeneration characterized by alterations in membranes, characteristic of cellular blebbing seen in apoptosis, a finding consistent with the visual observations made in the present study. In addition, cell shrinkage, and detachment were also observed.
The concentrations of Thimerosal inducing significant neuronal cell toxicity in the present study, as well as in several of the aforementioned studies, are compatible with physiological levels known to be induced by fetal and early infant exposure to Hg from Thimerosal-containing biologics and vaccines (Burbacher et al. 2005; Orct et al. 2006). For example, Burbacher et al. (2005) conducted a study in infant monkeys to toxicologically evaluate the distribution of Hg following the range of estimated doses of Thimerosal received by human infants receiving vaccines during the first six months of life. These researchers demonstrated that Thimerosal exposure resulted in post-dosing maximal brain total Hg content of 40–50 ng g 1. In addition, a significant 16 ng g 1 inorganic Hg fraction was found post-dosing that was observed to show no significant decline more than 120 days later. These levels of Hg in the brain are comparable to in vitro levels of Thimerosal shown to rapidly induce significant neuronal cytotoxicity in the present study, as well as that of Parran et al. (2005), who found an LC50 at 48 h for human neuroblastoma cell death of 0.87 ng g 1 Hg with Thimerosal incubation. Furthermore, the concentrations of Thimerosal inducing significant neuronal cell toxicity in the present study, as well as in several of the aforementioned studies, are also compatible with levels of Thimerosal exposure known to induce autistic-like behaviors and pathophysiology in a susceptible mouse model system (Hornig et al. 2004; Laurente et al. 2007). Other researchers have now demonstrated similar adverse effects in other animal models following low-concentration MeHg exposure (Falluel-Morel et al. 2007; Montgomery et al. 2008).
Autism pathophysiology
Additionally, the effects induced by Thimerosal in the present study, as well as from previous studies (Deth et al. 2008; Herdman et al. 2006; Humphrey et al. 2005; James et al. 2005; Parran et al. 2005; Waly et al. 2004; Yel et al. 2005), are consistent with recently emerging evidence documenting the brain pathophysiology present in patients diagnosed with ADs. The present study, as well as that of James et al. (2005), showed that human neuroblastoma cells were significantly more susceptible to Thimerosal-induced damage than human astrocytoma cells. Further, Toimela and Tahti (2004) evaluated co-cultures of neuroblastoma and astrocytoma cells following organic and inorganic Hg exposures. In co-cultures of neuroblastoma and astrocytoma cells, researchers visually observed that neuroblastoma cells were significantly damaged at Hg concentrations, which had little or no adverse effects on the astrocytoma cells. Consistent with these observations, Lopez-Hurtado and Prieto (2008) found striking differences in the density of glial cells, the density of neurons and the number of lipofuscin-containing neurons in brain regions associated with the production and processing of speech when patients diagnosed with ADs were compared to controls. Specifically, it was observed that the brains in the patients diagnosed with ADs had significantly greater mean densities of glial cells in comparison to controls. By contrast, the density of neurons was significantly decreased in the brain samples of patients diagnosed with autistic disorders in comparison with controls. It is important to note that visual images obtained from the brain samples of patients diagnosed with ADs were virtually identical in morphology with those observed in co-cultures of neuroblastoma and astrocytoma cells exposure to Hg by Toimela and Tahti (2004). Finally, these researchers observed that patients diagnosed with ADs had significantly increased numbers of lipofuscin-containing neurons comparison to controls. The presence of lipofuscin in neurons is significant with regard to Hg toxicity, because lipofuscin is a depot for heavy metals such as Hg (Opitz et al. 1996).
The present study, as well as several recent studies (Humphrey et al. 2005; James et al. 2005; Yel et al. 2005; Herdman et al. 2006), suggests that mitochondrial dysfunction and impaired oxidative–reduction are significant mechanisms underlying neuronal cell damage induced by Thimerosal. Consistent with these observations, studies identified evidence for mitochondrial dysfunction and impaired oxidative–reduction in patients diagnosed with ADs (Chauhaun and Chauhaun 2006). Chauhaun and Chauhaun (2006) postulated the overall pathogenesis as a condition resulting from environmental factors including Hg coupled with decreased levels of key body antioxidants, such as low glutathione or antioxidant enzymes leading to the enhanced production of free radicals. These studies showed that the resultant enhanced free-radical production may produce an increase in lipid peroxidation, protein oxidation, and DNA oxidation, all leading to enhanced oxidative stress. Further, it was found that the resultant rise in free radical production increased mitochondrial damage, impaired energy production, and enhanced excitotoxicity, all leading to increased oxidative stress. In considering the effects of oxidative stress on the mechanisms that mediate neuronal dysfunction and clinical symptoms in autism, enhanced oxidative stress was shown to result in impaired neuronal development, increased inflammatory response, impaired energy production, cell death, and decreased synaptic efficiency. Data indicated that these combined effects lead directly to the pathogenesis and clinical presentation of ADs.
Relative toxicities of the metal compounds tested
The purpose of the present study was also to compare Thimerosal-associated cellular damage with other potential environmental exposures to toxic metal compounds. When assessing the toxicity of the metal compounds tested on human neuroblastoma cells, repeatedly Thimerosal was observed to induce significantly greater mitochondrial and visually observed cellular damage than the other compounds tested. In evaluating the basis for the relative toxicity of the metal compounds examined, the following criteria appear to dictate the measured toxicity (the more criteria met, the greater the toxicity): (1) solubility of the compound in aqueous solutions; (2) solubility of the compound in lipid membranes; and (3) the relative intracellular toxicity of the compound. Thimerosal as a compound, based upon testing from the present study, appeared to excel in each of these criteria.
In considering each criteria for the compounds tested in the present study, the first criteria (i.e. aqueous solubility) was met by each compound tested, although Thimerosal does have extraordinary capacity to dissolve at a 1 : 1 concentration with water. The second criteria (i.e. lipid solubility) was effectively met by the short chained alkylmercurial compounds tested in the present study, including Thimerosal, in the form of its EtHg breakdown component, and by MeHg hydroxide. In contrast, Hg chloride, Pb acetate, and Al sulfate have limited lipid solubility. Finally, the third criterion (i.e. intracellular toxicity) was met most effectively by Thimerosal. The EtHg breakdown product of Thimerosal is known to intracellularly release inorganic Hg in the form of mercuric ions (Hg2þ) to a much greater extent than MeHg hydroxide (Fang and Fallen 1974; Suzuki et al. 1973). Mercuric ions have a significant affinity for intracellular proteins containing sulfhydryl groups, many of which are very important to cellular function (Elferink 1999). In contrast, MeHg hydroxide has a significantly lower affinity for sulfhydryl groups. Moreover, since MeHg hydroxide is more stable, it possesses a greater probability to re-cross the cellular membrane and leave the cell before it is metabolized, as opposed to Thimerosal’s EtHg metabolites. The ranking of cellular toxicity among the metals tested in the present study confirms that observed by several other researchers. For example, studies showed that the toxicity of Al5Pb5Hg chloride5Thimerosal to neuroblastoma cells (Deth et al. 2008; Waly et al. 2004). Other investigations also similarly reported that Thimerosal induced significantly greater cellular toxicity than MeHg (Parran et al. 2005; Yole et al. 2007).
Conclusion
The present study showed that Thimerosal-induced cellular damage among in vitro human neuronal and fetal model systems in a concentration- and time-dependent fashion. Thimerosal at low nanomolar concentrations was able to induce significant cellular toxicity in human neuron and fetal cells. Thimerosal-induced cellular cytotoxicity similar to that observed in pathophysiological studies of patients diagnosed with ADs. Namely, in both cases, there was evidence of mitochondrial dysfunction, reduced cellular oxidative– reduction activity, cell death, and cell degeneration. The present study also revealed that Thimerosal is significantly more toxic than several other well-established neurodevelopmental toxins. Finally, future studies should be conducted to further evaluate additional mechanisms for Thimerosal-induced cellular damage and to further assess potential co-exposures that may work to ameliorate or enhance its toxicity.
Acknowledgments
We wish to thank Lisa Sykes for help in reviewing this study. This study was funded by the nonprofit CoMeD, Inc. and by the nonprofit Institute of Chronic Illnesses, Inc. through a grant from the Brenen Hornstein Autism Research & Education Foundation. David Geier, Dr. Mark Geier, and Dr. Paul G. King have been involved in vaccine/biologic litigation.
References
Bigham, M., and R. Copes. 2005. Thiomersal in vaccines: Balancing the risk of adverse effects with the risk of vaccine-preventable disease. Drug Safety: An International Journal of Medical Toxicology and Drug Experience 28: 89–101.
Burbacher, T.M., D.D. Shen, N. Liberato, K.S. Grant, E. Cernichiari, and T.W. Clarkson. 2005. Comparison of blood and brain mercury levels in infant monkeys exposed to methylmercury or vaccines containing Thimerosal. Environmental Health Perspectives 113: 1015–21.
Chauhaun, A., and V. Chauhaun. 2006. Oxidative stress in autism. Pathophysiology: The Official Journal of the International Society for Pathophysiology/ISP 13: 171–81.
Chrysochoou, C., C. Rutishauser, C. Rauber-Luthy, T. Neuhaus, E. Boltshauser, and A. Superti-Furga. 2003. An 11 month-old boy with psychomotor regression and auto-aggressive behaviour. European Journal of Pediatrics 162: 559–61.
Collaborative on Health and the Environment’s Learning and Developmental Disabilities Initiative 2008. LDDI Scientific consensus statement on environmental agents associated with neurodevelopmental disorders. 1–35.
Counter, S.A., L.H. Buchanan, F. Ortega, and G. Laurell. 2002. Elevated blood mercury and neurootological observations in children of the Ecuadorian gold mines. Journal of Toxicology and Environmental Health A 65: 149–63.
Deth, R., C. Muratore, J. Benzecry, V.A. Power-Charnitsky, and M. Waly. 2008. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology 29: 190–201.
Elferink, J.G. 1999. Thimerosal: A versatile sulfhydryl reagent, calcium mobilizer, and cell functionmodulating agent. General Pharmacology 33: 1–6.
Environmental Working Group 2004. Overloaded?. New science, new insights about mercury and autism in susceptible children. Washington, DC: EWG Action Fund.
Fang, S.C., and E. Fallin. 1974. Uptake and subcellular cleavage of organomercury compounds by rat liver and kidney. Chemico–Biological Interactions 9: 57–64.
Falluel-Morel, A., K. Sokolowski, H.M. Sisti, X. Zhou, T.J. Shors, and E. Dicicco-Bloom. 2007. Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. Journal of Neurochemistry 103: 1968–81.
Geier, D.A., and M.R. Geier. 2006a. A meta-analysis epidemiological assessment of neurodevelopmental disorders following vaccines administered from 1994 through 2000 in the United States. Neuro Endocrinology Letters 27: 401–13.
Geier, D.A., and M.R. Geier. 2006b. A prospective assessment of porphyrins in autistic disorders: A potential marker for heavy metal exposure. Neurotoxicity Research 10: 57–64.
Geier, D.A., and M.R. Geier. 2006c. An evaluation of the effects of thimerosal on neurodevelopmental disorders reported following DTP and Hib vaccines in comparison to DTPH vaccine in the United States. Journal of Toxicology and Environmental Health A 69: 1481–95.
Geier, D.A., and M.R. Geier. 2007a. A case series of children with apparent mercury toxic encephalopathies manifesting with clinical symptoms of regressive autistic disorders. Journal of Toxicology and Environmental Health A 70: 837–51.
Geier, D.A., and M.R. Geier. 2007b. A prospective study of mercury toxicity biomarkers in autistic spectrum disorders. Journal of Toxicology and Environmental Health A 70: 1723–30.
Geier, D.A., and M.R. Geier. 2007c. A prospective study of thimerosal-containing Rho(D)-immune globulin administration as a risk factor for autistic disorders. The Journal of Maternal-Fetal & Neonatal Medicine: The Official Journal of the European Association of Perinatal Medicine, The Federation of Asia and Oceania Perinatal Societies, The International Society of Perinatal Obstetricians 20: 385–90.
Geier, D.A., E. Mumper, B. Gladfelter, L. Coleman, and M.R. Geier. 2008. Neurodevelopmental disorders, maternal rh-negativity, and Rho(D) immune globulins: A multi-center assessment. Neuro Endocrinology Letters 29: 272–80.
Geier, D.A., L.K. Sykes, and M.R. Geier. 2007. A review of Thimerosal (Merthiolate) and its ethylmercury breakdown product: Specific historical considerations regarding safety and effectiveness. Journal of Toxicology and Environmental Health. Part B, Critical Reviews 10: 575–96. Herdman, M.L., A. Marcelo, Y. Huang, R.M. Niles, S. Dhar, and K.K. Kiningham. 2006. Thimerosal induces apoptosis in a neuroblastoma model via the cJun N-terminal kinase pathway. Toxicological Sciences: An Official Journal of the Society of Toxicology 92: 246–53.
Hornig, M., D. Chian, and W.I. Lipkin. 2004. Neurotoxic effects of postnatal Thimerosal are mouse strain dependent. Molecular Psychiatry 9: 833–45.
Humphrey, M.L., M.P. Cole, J.C. Pendergrass, and K.K. Kiningham. 2005. Mitochondrial mediated Thimerosal-induced apoptosis in a human neuroblastoma cell line (SK-N-SH). Neurotoxicology 26: 407–16.
James, S.J., W. Slikker, S. Melnyk, E. New, M. Pogribna, and S. Jernigan. 2005. Thimerosal neurotoxicity is associated with glutathione depletion: Protection with glutathione precursors. Neurotoxicology 26: 1–8.
Kern, J.K., and A.M. Jones. 2006. Evidence of toxicity, oxidative stress, and neuronal insult in autism. Journal of Toxicology and Environmental Health. Part B, Critical Reviews 9: 485–99. Laurente, J., F. Remuzgo, B. Avalos, J. Chiquinta, B. Ponce, R. Avendano, and L. Maya. 2007. Neurotoxic effects of Thimerosal at vaccine doses on the encephalon and development in 7 days-old hamsters. Anales de la Facultad de Medicina, Universidad Nacional Mayor de San Marcos 68: 222–37.
Lopez-Hurtado, E., and J.J. Prieto. 2008. A microscopic study of language-related cortex in autism. American Journal of Biochemistry and Biotechnology 4: 130–45.
Marques, R.C., J.V. Bernardi, J.G. Dorea, W.R. Bastos, and O. Malm. Principal component analysis and discrimination of variables associated with pre- and post-natal exposure to mercury. International Journal of Hygiene and Environmental Health, In press.
Montgomery, K.S., J. Mackey, K. Thuett, S. Ginestra, J.L. Bizon, and L.C. Abbott. 2008. Chronic, low-dose prenatal exposure to methylmercury impairs motor and mnemonic function in adult C57/B6 mice. Behavioural Brain Research, 191: 55–61.
Mutter, J., J. Naumann, and C. Guethlin. 2007. Comments on the article ‘the toxicology of mercury and its chemical compounds’ by Clarkson and Magos (2006). Critical Reviews in Toxicology 37: 537–49.
Mutter, J., J. Naumann, R. Schneider, H. Walach, and B. Haley. 2005. Mercury and autism: Accelerating evidence? Neuro Endocrinology Letters 26: 439–46.
Nelson, B.K. 1991. Evidence for behavioral teratogenicity in humans. Journal of Applied Toxicology: JAT 11: 33–7.
Opitz, H., F. Schweinsberg, T. Grossmann, M.F. Wendt-Gallitelli, and R. Meyermann. 1996. Demonstration of mercury in the human brain and other organs 17 years after metallic mercury exposure. Clinical Neuropathology 15: 139–44.
Orct, T., M. Blanusa, M. Lazarus, V.M. Varnai, and K. Kostial. 2006. Comparison of organic and inorganic mercury distribution in suckling rat. Journal of Applied Toxicology: JAT 26: 536–9.
Palmer, R.F., S. Blanchard, S. Stein, D. Mandell, and C. Miller. 2006. Environmental mercury release, special education rates, and autism disorder: An ecological study of Texas. Health & Place 12: 203–9.
Palmer, R.F., S. Blanchard, and R. Wood. Proximity to point sources of environmental mercury release as a predictor of autism. Health & Place, In press.
Parran, D.K., A. Barker, and M. Ehrich. 2005. Effects of Thimerosal on NGF signal transduction and cell death in neuroblastoma cells. Toxicological Sciences: An Official Journal of the Society of Toxicology 86: 132–40.
Rury, J. 2006. Links between environmental mercury, special education and autism in Louisiana. PhD Thesis, Department of Environmental Studies, Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College.
Suzuki, T., T.L. Takemoto, H. Kashiwazaki, and T. Miyama. 1973. Metabolic fate of ethylmercury salts in man and animals. In Mercury, mercurials, mercaptans, ed. M.W. Miller and T. W. Clarkson, 209–40. Springfield, IL: Charles C. Thomas.
Tan, M., and J.E. Parkin. 2000. Route of decomposition of Thiomersal (Thimerosal). International Journal of Pharmaceutics 208: 23–34.
Toimela, T., and H. Tahti. 2004. Mitochondrial viability and apoptosis induced by aluminum, mercuric mercury and methylmercury in cell lines of neural origin. Archives of Toxicology 78: 565–74.
US National Institute of Environmental Health Sciences 1999. A research-oriented framework for risk assessment and prevention of children’s exposure to environmental toxicants. Environmental Health Perspectives 107: 510.
Waly, M., H. Olteanu, R. Banerjee, S.W. Choi, J.B. Mason, B.S. Parker, S. Sukumar, S. Shim, et al. 2004. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: A target for neurodevelopmental toxins and Thimerosal. Molecular Psychiatry 9: 358–70.
Windham, G.C., L Zhang, R. Gunier, L.A. Croen, and J.K. Grether. 2006. Autism spectrum disorders in related to distribution of hazardous air pollutants in the San Francisco Bay area. Environmental Health Perspectives 114: 1438–44.
Yel, L., L.E. Brown, K. Su, S. Gollapudi, and S. Gupta. 2005. Thimerosal induces neuronal cell apoptosis by causing cytochrome c and apoptosis-inducing factor release from mitochondria. International Journal of Molecular Medicine 16: 971–7.
Yole, M., M. Wickstrom, and B. Blakley. 2007. Cell death and cytotoxic effects in YAC-1 lymphoma cells following exposure to various forms of mercury. Toxicology 243: 40–57.
Young, H.A., D.A. Geier, and M.R. Geier, 2009. Thimerosal exposure in infants and neurodevelopmental disorders: An assessment of the computerized medical records in the Vaccine Safety Datalink. Journal of the Neurological Sciences, In press.
Zahir, F., S.J. Rizwi, S.K. Haw, and R.H. Khan. 2005. Low dose mercury toxicity and human health. Environmental Toxicology and Pharmacology 20: 351–60.
_______________
Notes:
a. Institute of Chronic Illnesses, Inc., Silver Spring, Maryland, USA;
b. CoMeD, Inc., Silver Spring, Maryland, USA;
c. The Genetic Centers of America, Silver Spring, Maryland, USA
1. Corresponding author. Email: [email protected] ISSN 0277–2248 print/ISSN 1029–0486 online © 2009 The Author(s). Published by Taylor & Francis. This is an Open Access article. Non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly attributed, cited, and is not altered, transformed, or built upon in any way, is permitted. The moral rights of the named author(s) have been asserted. DOI: 10.1080/02772240802246458 http://www.informaworld.com