by June M. Chan, Meir J. Stampfer, Edward Giovannucci, Peter H. Gann, Jing Ma, Peter Wilkinson, Charles H. Hennekens, Michael Pollak http://www.sciencemag.org SCIENCE z VOL. 279 z 23 JANUARY 1998 563
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.
Insulin-like growth factor–I (IGF-I) is a mitogen for prostate epithelial cells. To investigate associations between plasma IGF levels and prostate cancer risk, a nested case-control study within the Physicians’ Health Study was conducted on prospectively collected plasma from 152 cases and 152 controls. A strong positive association was observed between IGF-I levels and prostate cancer risk. Men in the highest quartile of IGF-I levels had a relative risk of 4.3 (95 percent confidence interval 1.8 to 10.6) compared with men in the lowest quartile. This association was independent of baseline prostate-specific antigen levels. Identification of plasma IGF-I as a predictor of prostate cancer risk may have implications for risk reduction and treatment.
***
The cell proliferation rate is positively correlated with the risk of transformation of certain epithelial cells (1). Insulin-like growth factors have mitogenic and antiapoptotic effects on normal and transformed prostate epithelial cells (2–4). Most circulating IGF-I originates in the liver, but IGF bioactivity in tissues is related not only to circulating IGF and IGF binding protein (IGFBP) levels, but also to local production of IGFs, IGFBPs, and IGFBP proteases (5). Person-to-person variability in levels of plasma IGF-I and IGFBP-3 [the major circulating IGFBP (5)] is considerable (6, 7), and plasma IGF-I levels appear to reflect heterogeneity in tissue IGF-I bioactivity (8–11).
To examine the potential relation between plasma IGF-I, IGF-II, and IGFBP-3 levels and prostate cancer risk, we conducted a prospective case-control study of men participating in the Physicians’ Health Study (12). At the start of the study (1982), the men (aged 40 to 82) provided medical information via mailed-in questionnaires, and 14,916 (68%) also provided plasma (12). Through 1992 follow-up was over 99% complete. Reports of prostate cancer were verified by medical records (13).
Cases and controls were selected from the 14,916 physicians who provided plasma. By March 1992, we confirmed 520 cases, of whom 152 had adequate volume for IGF assays in 1997. Levels of plasma steroid hormones (14), prostate-specific antigen (PSA) (15), and carotenoids, and CAG polymorphisms of the androgen receptor gene (16) had previously been measured in the same samples (17). On average, 7 years (minimum 5 6 months, maximum 5 9.5 years) elapsed between plasma collection and diagnosis.
We selected controls at random from men who provided blood and had not reported a diagnosis of prostate cancer up to the diagnosis date of the case. We excluded men with inadequate sample volume and who had total or partial prostatectomies by the time of the case diagnosis. We matched one control to each case on the basis of smoking (never, past, or current), duration of follow-up, and age within 1 year.
IGF-I, IGF-II, and IGFBP-3 were assayed by enzyme-linked immunoabsorbent assay (ELISA) with reagents from Diagnostic Systems Laboratory (Webster, Texas) (18, 19). A single IGF-I measurement is generally representative of levels over time (20, 21). We used paired t tests to compare the means of IGF-I, IGF-II, and IGFBP-3 levels between cases and controls. We examined age-standardized (using five groups: 40 to 50, 51 to 55, 56 to 60, 61 to 65, and 66 to 80) mean values of various predictors for prostate cancer within quartiles of IGF-I among the controls. Conditional logistic regression was used to analyze the associations between IGF and prostate cancer, after adjustment for other possible risk factors— PSA, height, weight, body mass index, CAG polymorphisms of the androgen receptor gene, and plasma levels of lycopene, estrogen, testosterone (T), dihydrotestosterone (DHT), sex hormone binding globulin (SHBG), prolactin, and 3aandrostanediol glucuronide (AAG) (14–16, 22–24). In view of the growth-inhibitory properties of IGFBP-3 and its potential to reduce the bioactivity of IGF (25), we hypothesized that high levels of IGFBP-3 would be inversely related to risk. Because levels of IGF-I and IGFBP-3 were highly correlated, it was necessary to simultaneously adjust for these factors in regression models to observe their independent effects.
We estimated relative risks (RRs) from the odds ratios and computed 95% confidence intervals (CIs) (24). In stratified analyses, we used unconditional logistic regression models and adjusted for age (eight 5-year categories) and smoking (never, past, and current) in the models to make full use of the data without restriction to the matched pairs (24). We also separately examined high grade/stage cases, low grade/ stage cases, and cases occurring after the first 5 years of follow-up.
Because PSA acts as an IGFBP protease in prostatic tissue (26), we investigated possible interactions involving PSA. We classified men by quartile of IGF-I and low (#4 ng/ml) versus high (.4 ng/ml) PSA level, creating eight categories of IGF-I and PSA. Similar methods were used to examine potential interactions between IGF-I and plasma androgens (with the controls’ medians as cutpoints for low and high androgen levels).
All exposures of interest and covariates, with the exceptions of CAG repeats (dichotomized), age, and smoking, were analyzed in quartile groups (based on controls’ distributions) with the lowest quartile as the reference category. We tested linear trends for statistical significance by assigning the medians of each quartile as scores (24).
The mean level of IGF-I among the cases (269.4 ng/ml) was significantly higher than among controls (248.9 ng/ml) (P 5 0.03). Means of IGF-II and IGFBP-3 were similar among cases and controls (P 5 0.85 and 0.95, respectively). Table 1 presents age-standardized means of IGF-II, IGFBP-3, lycopene, estrogen, T, DHT, SHBG, weight, height, body mass index, and medians of PSA among 152 control men, within quartiles of IGF-I. PSA and estrogen had weak positive associations with IGF-I levels, and lycopene levels were slightly lower among men in the highest quartile of IGF-I. There was no significant correlation between IGF-I and any of these factors except IGF-II (r 5 0.5) and IGFBP-3 (r 5 0.6).
IGF-I was significantly associated with prostate cancer risk in a univariate analysis; men in the highest quartile had an RR of 2.4 (95% CI 1.2 to 4.7) as compared with men in the lowest quartile (Table 2). With further adjustment for IGFBP-3, these men had more than four times the risk of prostate cancer compared with men in the reference group (RR 5 4.3, 95% CI 1.8 to 10.6). IGF-II and IGFBP-3 were not associated with prostate cancer risk when examined individually, but IGFBP-3 was inversely associated with risk after controlling for IGF-I (RR for fourth versus first quartile 0.4, 95% CI 0.2 to 1.0). This suggests that the inverse association with IGFBP-3 in the univariate analysis was masked because of its high correlation with IGF-I; the multivariate model shows the independent effects of each of these factors. There was a significant linear trend between IGF-I and prostate cancer risk, especially after adjustment for IGFBP-3; a 100 ng/ml increase in IGF-I corresponded to an approximate doubling of risk (RR 5 2.1 per 100 ng/ml increase, 95% CI 1.3 to 3.2).
IGF-I remained a significant independent predictor of prostate cancer risk even after inclusion of quartiles of weight, height, body mass index, lycopene, androgen receptor CAG repeats, and plasma hormone levels (estrogen, T, DHT, SHBG, prolactin, and AAG) in the multivariate models. Adding quartiles of PSA to the model attenuated the association for IGF-I slightly, although the results remained significant (RR 5 3.3, 95% CI 1.1 to 10.1 for the fourth versus first quartile, after adjustment for IGFBP-3). This underestimates the effect of IGF-I because controlling for PSA levels partly controls for the presence of undiagnosed prostate cancer.
To investigate whether the associations between IGF-I and prostate cancer could be due to increased IGF-I levels among preclinical undiagnosed cases in 1982, we repeated the basic analyses including only those men who were diagnosed 5 years or more after the start of follow-up. With the remaining 125 cases and 152 controls, we observed similar results to those from previous analyses with all cases and controls. The effect of IGF-I adjusted for PSA was also unchanged.
Comparison of the association between IGF-I and prostate cancer risk among men with high grade/stage versus low grade/stage cancer at diagnosis revealed no significant difference [RR for the fourth versus first quartile of IGF-I 5 3.4 (95% CI 1.1 to 10.1) for high grade/stage and 5.5 (95% CI 1.9 to 15.5) for low grade/stage cancers].
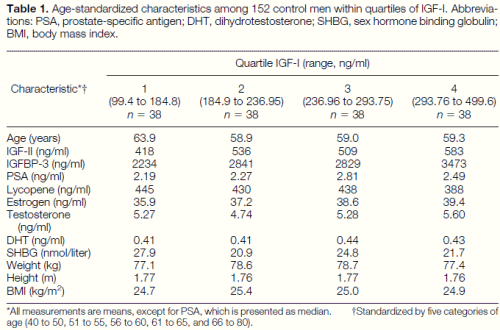
Table 1. Age-standardized characteristics among 152 control men within quartiles of IGF-I. Abbreviations: PSA, prostate-specific antigen; DHT, dihydrotestosterone; SHBG, sex hormone binding globulin; BMI, body mass index. *All measurements are means, except for PSA, which is presented as median. †Standardized by five categories of age (40 to 50, 51 to 55, 56 to 60, 61 to 65, and 66 to 80).
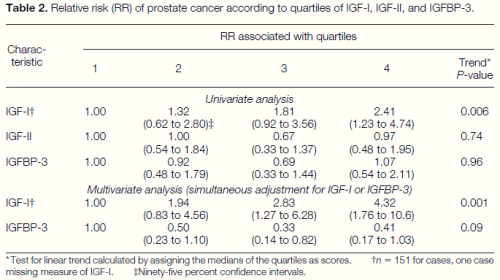
Table 2. Relative risk (RR) of prostate cancer according to quartiles of IGF-I, IGF-II, and IGFBP-3. * Test for linear trend calculated by assigning the medians of the quartiles as scores. †n 5 151 for cases, one case missing measure of IGF-I. ‡Ninety-five percent confidence intervals.
When we stratified subjects by the median case baseline age of 60, the increased risk associated with IGF-I was stronger among the older men. Men over age 60 and in the highest quartile of IGF-I had an RR of 7.9 (95% CI 2.1 to 30.7) with adjustment for IGFBP-3, compared with men of similar age and in the lowest quartile; the comparable RR among men age 60 or less was 1.6 (95% CI 0.4 to 6.1). There was a significant linear trend, however, among both older (P 5 0.006) and younger (P 5 0.047) men, and a formal test for interaction between age and IGF-I was not significant (P 5 0.43). We also examined IGF-I within strata of smoking and within strata of six plasma hormones but observed no evidence of interaction.
As previously reported (15), men with elevated baseline PSA levels were more likely to be subsequently diagnosed with prostate cancer than those with PSA levels less than or equal to 4 ng/ml (Table 3). However, plasma IGF-I level was strongly related to risk of developing prostate cancer even among men with a baseline PSA level less than or equal to 4 ng/ml (the multivariate RR of clinical diagnosis during followup increased from 1.0 to 4.6 across quartiles of IGF-I, adjusted for IGFBP-3, age, and smoking). Given that men with PSA levels greater than 4 ng/ml have a high likelihood of harboring occult prostate cancer (15), these data suggest a possible effect of IGF-I on the natural history of clinically occult prostate cancer (the multivariate RR increased from 3.9 to 17.5 across quartiles of IGF-I). Men in the highest quartile of IGF-I had ;4.5 times greater risk than men in the lowest quartile regardless of their PSA levels; hence, a combined assessment of IGF-I and PSA levels may better predict prostate cancer than a PSA measure alone. These results were unchanged when we excluded cases occurring during the first 5 years of follow-up.
Our data support the hypothesis that higher plasma IGF-I levels are associated with higher rates of malignancy in the prostate gland. It is possible that other measures of IGF-I physiology (for example, adolescent or early adulthood mean IGF-I assessed over time, tissue IGF bioactivity, or rate of cell turnover in the prostate gland) would better capture the true etiologically relevant variable. To the extent that our single measurement is a proxy for such a variable and that the measurement errors are nonsystematic and proportionately equal among cases and controls, we have reduced the observable variation between our cases and controls, and our results may underestimate the true association between IGF-I and prostate cancer risk (24). Measurement error in assessing prostate cancer outcome is minimal given the physician study base and the histologic confirmation of all cases, and any underascertainment of existing cases would not bias the relative risks in our study (24).
A small case-control study (n 5 52 cases), in which blood samples were drawn from men already diagnosed with prostate cancer and healthy controls, observed a multivariate relative risk of prostate cancer of 1.9 per 60 ng/ml increase in IGF-I (95% CI 1.0 to 3.7) (27). However, the retrospective design used in that study could not rule out an effect of the cancer on IGF-I levels.
The association between circulating IGF-I level and risk of prostate cancer is stronger than that of any previously reported risk factor, including steroid hormone levels (14) or anthropomorphic variables (22, 23, 28–30). Previous reports showing a weak relation between prostate cancer risk and height (22, 28, 29) are of particular interest in the context of our results, as IGF-I levels have been reported to be loosely correlated with height (6), and height may act as a weak surrogate for IGF-I. Plasma IGF-I level, in turn, may be related to risk because it represents a determinant of or a surrogate for prostate tissue IGF bioactivity, which affects cellular proliferation rate. In this study population, height was moderately associated with prostate cancer risk, independent of weight, age, smoking, IGF-I, and IGFBP-3 (RR 5 1.1 per centimeter increase in height, P 5 0.05). However, we did not observe an association between IGF-I or IGFBP-3 and height in this study, possibly as a result of small sample size or older age of the subjects. A small study (n 5 21 cases) that found high birth weight to be associated with a higher incidence of prostate cancer may also be consistent with the observed effect of IGF (30), as birth weight may be positively correlated with IGF-I level (31).
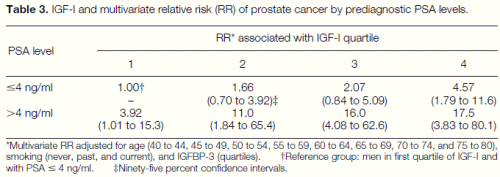
Table 3. IGF-I and multivariate relative risk (RR) of prostate cancer by prediagnostic PSA levels. *Multivariate RR adjusted for age (40 to 44, 45 to 49, 50 to 54, 55 to 59, 60 to 64, 65 to 69, 70 to 74, and 75 to 80), smoking (never, past, and current), and IGFBP-3 (quartiles). †Reference group: men in first quartile of IGF-I and with PSA # 4 ng/ml. ‡Ninety-five percent confidence intervals.
Reduction of androgen action has been the principal strategy under investigation for prostate cancer prevention. If our results are confirmed, pharmacological approaches to decreasing IGF-I bioactivity may warrant investigation as risk-reduction strategies specifically targeted at men at high risk due to increased IGF-I levels. Partial suppression of the growth hormone (GH)–IGF-I axis by somatostatin analogs (32) or GHreleasing hormone antagonists (33) are two possibilities. Finally, our results raise concern that administration of GH or IGF-I over long periods, as proposed for elderly men to delay the effects of aging (34), may increase risk of prostate cancer.
REFERENCES AND NOTES
1. S. M. Cohen and L. B. Ellwein, Science 249, 1007 (1990).
2. P. Cohen, D. M. Peehl, R. G. Rosenfeld, Hormone Metab. Res. 26, 81 (1994).
3. P. Cohen, D. M. Peehl, G. Lamson, R. G. Rosenfeld, J. Clin. Endocrinol. Metab. 73, 401 (1991).
4. R. Rajah, B. Valentinis, P. Cohen, J. Biol. Chem. 272, 12181 (1997).
5. J. I. Jones and D. R. Clemmons, Endocr. Rev. 16, 3 (1995).
6. A. Juul et al., J. Clin. Endocrinol. Metab. 78, 744 (1994).
7. A. Juul et al., ibid. 80, 2534 (1995).
8. Acromegaly and GH deficiency are examples where changes in tissues are correlated with serum IGF-I levels, implying a relation between serum IGF-I levels and tissue IGF-I bioactivity.
9. M. Pollak et al., J. Natl. Cancer Inst. 82, 1693 (1990).
10. H. Huynh, X. Yang, M. Pollak, J. Biol. Chem. 271, 1016 (1996).
11. H. Huynh, X. Yang, M. Pollak, Cell Growth Differ. 7, 1501 (1996).
12. Steering Committee of the Physicians’ Health Study Research Group, N. Engl. J. Med. 321, 129 (1989). The Physicians’ Health Study began in 1982 as a randomized, double-blinded, placebo-controlled trial of b-carotene and aspirin in 22,017 U.S. male physicians aged 40 to 82 without prior diagnosed cardiovascular disease or cancer. Each participant provided informed consent. Before randomization, the men had their blood drawn into vacutainer tubes containing EDTA (anti-coagulant). The blood was centrifuged and returned in polypropylene cryopreservation vials by overnight prepaid courier with cold packs to keep it cool. Upon receipt, the plasma was stored at 282°C. No specimen thawed or warmed substantially during storage.
13. The Whitmore-Jewett classification scheme was used to identify stage, and cases without pathological staging were considered indeterminate, unless there was evidence of metastases. Cases presenting as stage C or D were considered “high grade/ stage cancer.” Also, we assigned that category to stage A, B, or indeterminate cases with either poor histological differentiation or a Gleason score of 7 or higher.
14. P. H. Gann, C. H. Hennekens, J. Ma, C. Longcope, M. J. Stampfer, J. Natl. Cancer Inst. 88, 1118 (1996).
15. P. H. Gann, C. H. Hennekens, M. J. Stampfer, J. Am. Med. Assoc. 273, 289 (1995).
16. E. Giovannucci et al., Proc. Natl. Acad. Sci. U.S.A. 94, 3320 (1997).
17. Selection bias is minimal here as it is unlikely that subjects returned blood samples or provided adequate blood volume differentially on the basis of any relation between their IGF levels in 1982 and later development of prostate cancer. Previous study has shown that cases who did and did not provide blood samples were not appreciably different in their baseline lifestyle characteristics (15).
18. The IGF-I values obtained by the ELISA were highly correlated (Pearson r 5 0.97) with values obtained by radioimmunoassay after acid chromatography. All assays were carried out in a blinded manner, and quality control samples were included within assay runs. Average intra-assay coefficients of variation for IGF-I, IGF-II, and IGFBP-3 were 4.9%, 3.0%, and 9.0%, respectively.
19. To evaluate the effect of our blood collection methods on IGF-I levels, we compared IGF-I and IGFBP-3 levels in blood samples that were processed and serum frozen immediately after venipuncture (the usual collection and processing methods) to samples that were stored as heparinized whole blood for 24 and 36 hours before processing (mimicking our collection conditions). The mean IGF-I and IGFBP-3 values were almost identical and the interclass correlations between results of the two collection methods were 0.98 for IGF-I and 0.96 for IGFBP-3, indicating that our collection methods did not adversely affect sample integrity.
20. To examine how well a single measurement of IGF-I represents levels over time, we collected two blood samples each from 16 people, 8 weeks apart (time 1 and time 2). The correlation between blood levels taken at time 1 and time 2 was 0.65.
21. D. Goodman-Gruen and E. Barrett-Connor, Am. J. Epidemiol. 145, 970 (1997).
22. E. Giovannucci, E. B. Rimm, M. J. Stampfer, G. A. Colditz, W. C. Willett, Cancer Epidemiol. Biomarkers Prev. 6, 557 (1997).
23. S.-O. Andersson et al., J. Natl. Cancer Inst. 89, 385 (1997).
24. K. J. Rothman, Modern Epidemiology (Little, Brown, Boston, MA, 1986), pp. 82–97 and 286–310.
25. M. M. Rechler, Endocrinology 138, 2645 (1997).
26. P. Cohen et al., J. Clin. Endocrinol. Metab. 75, 1046 (1992).
27. C. S. Mantzoros et al., Br. J. Cancer 76, 1115 (1997).
28. C. La Vecchia et al., Int. J. Cancer 45, 275 (1990).
29. P. R. Hebert et al., Cancer Causes Control 8, 591 (1997).
30. G. Tibllin, M. Eriksoon, S. Cnattingius, A. Ekbom, Epidemiology 6, 423 (1995).
31. C. Lassarre et al., Pediatr. Res. 29, 219 (1991).
32. M. Pollak and A. Schally, Proc. Soc. Exp. Biol. Med. 217, 143 (1998).
33. M. Zarandi et al., Proc. Natl. Acad. Sci. U.S.A. 91, 12298 (1994).
34. D. Rudman et al., N. Engl. J. Med. 323, 1 (1990).
35. Supported by National Research Service Award 5 T32 CA 09001-20, National Cancer Institute grants CA-58684 and CA-42182, and the National Cancer Institute of Canada.
1 August 1997; accepted 4 December 1997
_______________
Notes:
J. M. Chan and M. J. Stampfer, Department of Epidemiology, Harvard School of Public Health, 677 Huntington Avenue, Boston, MA 02115, USA.
E. Giovannucci and J. Ma, Channing Laboratory, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA. P. H. Gann, Department of Preventive Medicine, Northwestern University Medical School, Chicago, IL 60611, USA.
P. Wilkinson and M. Pollak, Cancer Prevention Research Unit, Departments of Medicine and Oncology, Lady Davis Research Institute of the Jewish General Hospital and McGill University, Montreal, Canada H3T1E2.
C. H. Hennekens, Division of Preventive Medicine, Department of Medicine and Department of Ambulatory Care and Prevention, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115, USA.
*To whom correspondence should be addressed. E-mail: [email protected]