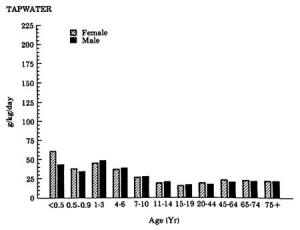
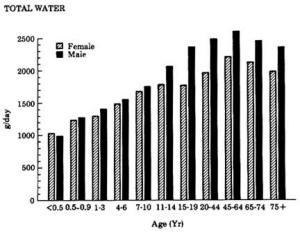
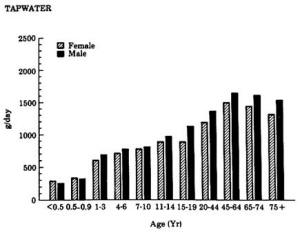
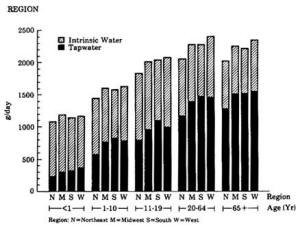
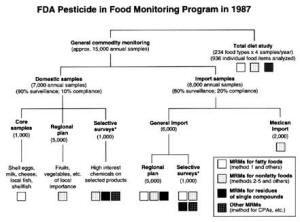
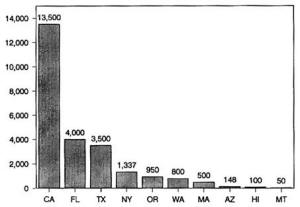
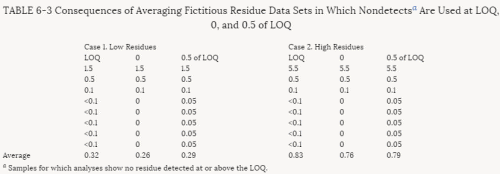
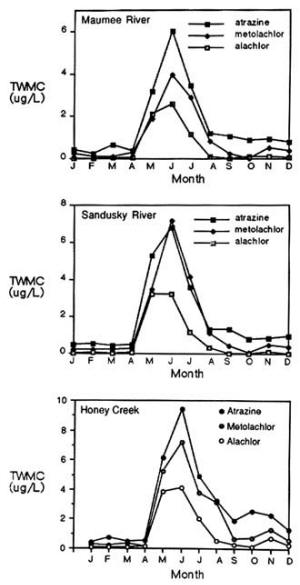
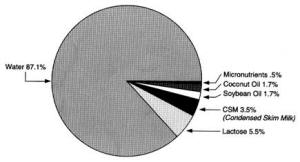
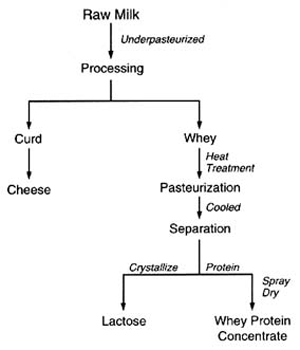
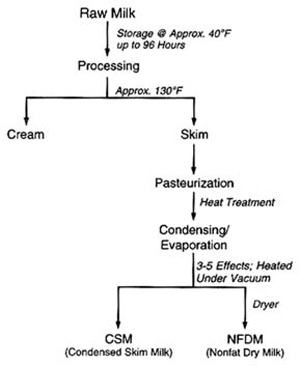
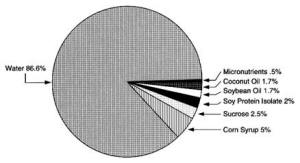
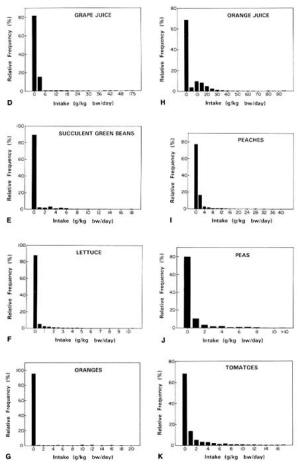
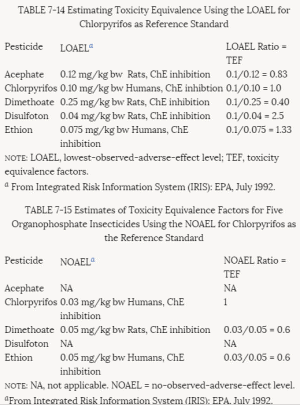
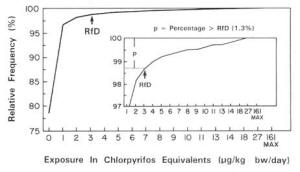
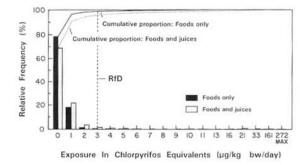
4. Methods for Toxicity Testing
THE PURPOSE OF THIS CHAPTER is to familiarize the reader with the testing that is currently conducted by a manufacturer prior to and during the process of submitting a petition to register a pesticide. Codified toxicologic evaluation of potential pesticides has been a requirement in the United States for approximately 50 years. The testing requirements and guidelines continue to evolve based on new science. This chapter identifies the current testing that is pertinent to the young animal and young human as well as aspects of testing that are needed to fill the data gaps to better ensure the protection of infants and children. The current testing guidelines can be found in Pesticide Assessment Guidelines issued by the Environmental Protection Agency (EPA, 1991a,b).
Data, including those derived from toxicity testing, crop residue analyses, environmental fate testing, and ecotoxicology testing, are generated by the manufacturer of a pesticide to meet the mandatory requirements of the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) for pesticide registration. Although these data are essential to the EPA's registration process, other data generated by EPA itself, as well as by other government institutions and academia, are considered in the registration decision-making process.
EPA has issued 194 registration standards on 350 chemicals used as active and inert ingredients in pesticide products. These standards are published by EPA and are intended to upgrade and update the data base on a previously registered pesticide or class of pesticide products. They call for additional studies in the areas of toxicity testing, crop residue analyses, environmental fate, and ecotoxicology testing. This testing must be conducted within an EPA-mandated time frame to allow for the continued registration of given product. The list of pesticides for which registration standards have been issued is referred to as List A and can be found in Appendix I of the Federal Register notice of February 22, 1989. Under the FIFRA Amendments of 1988, the data bases on the remaining registered pesticide products are being upgraded in five phases over a 9-year period.
TABLE 4-1 End Points for Various Toxicity Studies
Study / End Points
Developmental toxicity / Fetus: mortality, growth retardation, skeletal variations, gross external malformations, soft tissue/internal organ defects
Female parent: general toxicity
Reproductive Toxicity / Male parent: general toxicity, effects on fertility, reproductive organ changes
Offspring: effects on viability, sex ratio, growth, behavior
Carcinogenicity / Tumor development and general toxicity
Neurotoxicity / Behavior, function, and motor activity deficits; microscopic nervous tissue changes
Mutagenicity / Heritable lesions leading to altered phenotypes
SOURCE: EPA, 1984
The first sections of this chapter describe in detail the present toxicity testing procedures for pesticides in relation to their registered use patterns and EPA's proposed changes or additions to these procedures. The conclusions and recommendation of the committee for further changes and additions to the toxicity testing battery to allow for more adequate consideration of the special testing needs for infants and children are presented.
CURRENT METHODS: GENERAL CONSIDERATIONS
Toxicity studies are required to assess potential hazards to humans through the acute, subchronic, and chronic exposure of laboratory animals to pesticides. The more specific types of toxicity that are determined include carcinogenicity; developmental (including teratogenicity in offspring) and reproductive toxicity; mutagenicity; and neurotoxicity (Table 4-1). Detailed information on the metabolism or biotransformation of the pesticide is also obtained. Consideration is given to testing individual metabolites in animals, and in or on pesticide-treated plants to which humans could exposed through their diet. The extent of metabolite testing required depends on the level of potential toxicity and environmental persistence of the metabolite. With the exception of the acute toxicity tests, most tests are conducted to determine the nature of any toxicity that can be produced by repeatedly dosing animals over an extended period. The results enable toxicologists to estimate the safety of a material of humans (Loomis, 1978).
Weil (1972) published the following set of guidelines, which reflected a consensus among toxicologists. These should be considered before initiating a toxicity test:
1. Use, wherever practical or possible, one or more species that biologically handle the material qualitatively and/or quantitatively as similarly as possible to man. For this, metabolism, absorption, excretion, storage and other physiological effects might be considered.
2. Where practical, use several dose levels on the principle that all types of toxicologic and pharmacologic actions in man and animals are dose-related. The only exception to this should be the use of a single, maximum dosage level if the material is relatively nontoxic; this level should be a sufficiently large multiple of that which is attainable by the applicable hazard exposure route, and should not be physiologically impractical.
3. Effects produced at higher dose levels (within the practical limits discussed in 2) are useful for delineating mechanism of action, but for any material effect, some dose level exists for man or animal below which this adverse effect will not appear. This biologically insignificant level can and should be set by use of a proper uncertainty factor and competent scientific judgment.…
4. Statistical tests for significance are valid only on the experimental units (e.g., either litters or individuals) that have been mathematically randomized among the dosed and concurrent control groups….
5. Effects obtained by one route of administration to test animals are not a priori applicable to effects by another route of administration to man. The routes chosen for administration to test animals should, therefore, be the same as those to which man will be exposed. Thus, for example, food additives for man should be tested by admixture of the material in the diet of animals.
In general, Weil's guidelines are considered by EPA in its toxicity testing requirements and subsequent evaluation of results for pesticides. One exception to Weil's points is found in his guideline 3. EPA does not recognize the existence of a dose level at which a carcinogen will not exert its effect. For carcinogens, EPA generally accepts a risk of 10-6, as extrapolated from bioassays using the nonthreshold modification of the linearized multistage model of Armitage and Doll (1954), as adequate for the protection of humans.
The selection of animal species for toxicity tests depends on life span, behavior, availability, and overall costs. EPA recommends using rats for subchronic, chronic, carcinogenicity, and reproduction studies; mice for carcinogenicity studies; and dogs for subchronic and chronic studies. Rats are routinely used for acute oral and inhalation studies and rabbits for eye and skin irritation studies and acute dermal studies. One exception to this is the use of guinea pigs for dermal sensitization testing. The rat and rabbit are recommended for developmental toxicity (teratogenicity) testing. Justification must be provided for the use of species other than those outlined above.
The number of animals to be tested in each dose group depends on a number of factors, including the purpose of the experiment, the required sensitivity of the study, the reproductive capacity and the fertility of the species, economic aspects, and the availability of animals (IPCS, 1990). Table 4-2 lists the minimum number of animals required by EPA for some toxicity studies. For the most part, these numbers are consistent with those recommended by the International Program Chemical Safety (IPCS).
The selection of dose levels for subchronic studies should be based on the results of acute toxicity testing, on range-finding studies, and on pharmacokinetic (metabolism, including rate in various tissues) data. For subchronic studies, four dose groups of animals should be included: a control group; a low-dose group (a dose that produces no compound related toxicity); a mid-dose group (a dose that elicits some minimal signs of toxicity); and a high-dose group (a dose that results in toxic effects but not in an incidence of fatalities that would prevent a meaningful evaluation; for nonrodents, there should be no fatalities) (EPA, 1984). This same guidance is relevant to chronic toxicity and reproduction studies. For teratology studies, the highest dose tested should elicit some signs of maternal toxicity, but the toxicity should not obscure the results.
The one notable exception to this guidance pertains to carcinogenicity studies. The highest dose levels for these studies should be at a maximum tolerated dose (MTD), as determined in 90-day toxicity studies in the appropriate test species and from pharmacokinetic information on the material being tested. The Committee on Risk Assessment Methodology of the National Research Council (NRC) recently examined the criteria for the MTD and other doses used in carcinogenicity studies (NRC, 1993). The EPA has issued its own guidance for the selection of this dose level. Some of the factors to consider in selecting an MTD are: 10% decrement in body weight gain in 90-day study; observation of potential life-threatening lesions during microscopic examination of organs, e.g., liver necrosis; significant inhibition of cholinesterase activity in two biological compartments, such as brain and plasma; and significant signs of anemia or other biologically relevant effects on blood.

TABLE 4-2 Animal Model Requirements in Toxicity Studies
a Either males or females may be used in this test.
b The number of animals used depends on the method used. Several different experimental methods are acceptable.
c EPA prefers that one male rat be housed with one female during mating.
d Number of pregnant females required.
e 50 rats and 50 mice of each sex.
SOURCE: EPA, 1984.
In general, EPA has set a cap on dosing of 1.0 g/kg/day for toxicity tests other than acute studies. This dose level is referred to as the limit dose and corresponds to approximately 20,000 ppm in the diet of rats, 7,000 ppm in the diet of mice, and 40,000 ppm in the diet of dogs.
The duration of exposure for toxicity testing of a pesticide depends on the expected duration of human exposure to the pesticide in practice. The typical length of various toxicity tests and the number of doses administered are shown in Table 4-2. Repeated dosing refers to dosing once per day for the designated number of days. When the material is given to the test animals in their diet, dosing is usually continuous for 7 days a week. If the material is administered by gavage (oral bolus dose), by dermal application, or by inhalation, doses are frequently given 5 days a week, which is acceptable to EPA because of practical considerations (EPA, 1984).
The type of statistical analysis performed on the toxicity data resulting from these studies depends on the type of data under consideration (see, for example, Gad and Weil, 1982, for review). Interpreting the meaning of statistical significance for any particular parameter depends on the dose level at which it was achieved, the biological significance of the finding, and the normal spontaneous occurrence of this finding in the strain and species being tested.
For regulatory purposes, the no-observed-effect level (NOEL) is defined as a dose level at which no effects attributable to the pesticide under test can be found. A no-observed-adverse-effect level (NOAEL) can also be determined for each study; however, EPA does not routinely use the NOAEL to regulate pesticide usage. To establish a NOAEL, the toxicologist must determine what is and what is not adverse effect, which can be defined differently by different scientists. For example, effects such as hair loss can be considered adverse by some and not by others. Plasma and red blood cell cholinesterase inhibition can be viewed as either an adverse effect or simply as a market of exposure to a pesticide.
EPA uses the NOEL to calculate the acceptable daily intake (ADI) of the pesticide under consideration. More recently, the EPA has replaced the ADI with the reference dose, or RfD. Chronic studies, such as reproduction studies and lasting 1 year or longer in the rat or dog are used for this purpose. EPA does not routinely use the NOEL determined from teratology (developmental toxicity) studies for calculating ADIs because the observed effect are not considered chronic; however, these NOELs can be used to support the calculated ADI. EPA does routinely use developmental toxicity NOELs for other types of risk assessments, such as calculating the risk from acute, daily dietary or occupational exposure or from exposure of homeowners to a developmental toxicant.
EPA's toxicity testing requirements for food and nonfood use pesticides have been published in 40 CFR Part 158. In general, for food use chemical with maximum human exposure, the following toxicity tests are required:
• acute oral toxicity
• acute dermal toxicity
• acute inhalation toxicity
• primary eye irritation
• primary dermal irritation
• chronic feeding toxicity
• dermal sensitization
• acute neurotoxicity
• 90-day toxicity
• 21-day dermal toxicity
• 90-day neurotoxicity study
• reproduction study
• carcinogenicity
• developmental toxicity
• mutagenicity tests
• general metabolism study
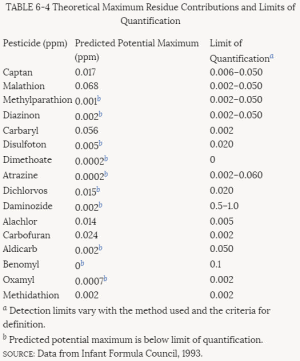
More than 30% of the tests for pesticides submitted to EPA in the past have been rejected. Those rejected must be resubmitted until they are in conformance with EPA criteria before registrations can be granted. The criteria for rejection are summarized in Table 4-3. Some of them fall in the category of regulatory policy; others involve scientific concerns. The most commonly cited reason for noncompliance is lack of characterization of the test material. To improve the quality of testing and incorporate new scientific methods in its testing requirements, EPA is currently revising the 40 CFR Part 158 data requirements for food and nonfood use pesticides. The proposed revisions to these requirements can be found in Table 4-4.
ACUTE TOXICITY STUDIES
General Description
Acute toxicity studies provide information on the potential for health hazards that may arise as result of short-term exposure. Determination of acute oral, dermal, and inhalation toxicity is usually the initial step in evaluating the toxic characteristics of a pesticide. In each of these tests the animal is exposed to the test material only once on 1 day. Together with information derived from primary eye and primary dermal irritation studies (also 1 dose on 1 day), which assess possible hazards resulting from pesticide contact with eyes and skin, these data provide a basis for precautionary labeling and may influence the classification of a pesticide for restricted use. Acute toxicity data also provide information used to determine the need for child-resistant packaging, for protective clothing requirements for applicator, and for calculation of farm worker reentry intervals. A minimum number of animals, usually adults, are used in these studies and only the end points of concern are monitored, i.e., mortality, observable skin or eye effects, dermal sensitization, and observable neurotoxic behavioral changes. One exception is the inclusion of microscopic examination of neural tissues in the newly required acute neurotoxicity study.
EPA's Proposed Changes
Guideline number 81-1 (EPA, 1984), acute oral study in the rat, would be revised to include special visual system testing, which would be required for all organophosphate pesticide and other pesticides known to affect the visual system.
TABLE 4-3 Summary of EPA Rejection Factors
Guideline / Rejection Factor
Acute Oral Toxicity (81-1) / Lack of characterization of the test material
Inadequate dose levels to calculate LD50
Acute Dermal Toxicity (81-2) / Lack of characterization of the test material
Inadequate percentage of body surface area exposed
No quality assurance statement
Improper number of animals tested per dose group
Only one sex tested
Omitted source, age, weight, or strain of test animal
Acute and 90-Day Inhalation (81-3 and 82-4) / Less than 25% of particles were <1 µm; LC50 could not be calculated; highest concentration did not produce toxicity
Inadequate reporting of exposure methodology
Protocol errors
Lack of characterization of the test material
Compound preparation
Chamber concentration not measured
Primary Eye Irritation (81-5) / Lack of characterization of the test material
Primary Dermal Irritation (81-5) / Lack of characterization of the test material
No quality assurance statement and/or no Good Laboratory Practice (GLP) statement
Improper test material application/preparation
Omitted source, age, weight, or strain of test animal
Missing individual/summary animal data
Dermal Sensitization (81-6) / Control problems
Dosing level problems
Lack of characterization of the test material
Unacceptable protocol or other protocol problems
Individual animal scorers or data missing
Scoring method or other scoring problem
Reporting deficiencies or no quality assurance statement
90-Day Feeding—Rodent (82-1(a)) / A NOEL was not established
Lack of characterization of the test material or incorrectly reported
Lack of clinical chemistry and/or lack of histopathology
90-Day Feeding—Nonrodents (82-1(b)) / Reporting deficiencies
Lack of characterization of the test material
A NOEL was not established
An investigation parameter missing
Guideline / Rejection Factor
90-Day Feeding—Nonrodents (cont.) (82-1(b)) / Information on the pilot study and other problems associated with dose level selection
An investigational parameter missing
Information on the pilot study and other problems associated with dose level selection
21-Day Dermal Toxicity (82-2) / Lack of characterization of the test material
Raw data analyses incomplete or missing
A systemic NOEL was not established
Inadequate percentage of body surface area exposed in each dose group
Insufficient number of dose levels tested
90-Day Dermal Toxicity (82-3) / Lack of characterization of the test material
A systemic NOEL was not established
Incomplete/missing raw animal data analyses
Insufficient number of dose levels tested
Poorly controlled test environment
Chronic Feeding/Carcinogenicity—Rats (82-3(a) and (83-2(a)) / Missing histopathology information
Missing information in study reports
MTD was not achieved
Missing historical control data
Lack of characterization of the test material
Deficiencies in reporting the study data
Carcinogenicity—Mice (83-2(b)) / Histopathology information missing
MTD was not achieved
Lack of historical control data
Information missing in study reports
Lack of characterization of the test material
Deficiencies in reporting of study data
Developmental Toxicity—Rodents (83-3(a)) / Missing historical controls
Lack of characterization of the test material
Information missing or requiring clarification of the laboratories' methods
Information missing or requiring clarification of the laboratories' results
A NOEL was not established
Statistical problems
Did not use conventional assessments for skeletal or visceral examinations
Developmental Toxicity—Nonrodents (83-3(b)) / Clarification of laboratory procedures of interpretation of the data
Individual maternal or fetal data missing
Missing historical controls
Lack of characterization of the test material
Excessive maternal toxicity
Guideline / Rejection factor
Developmental Toxicity—Nonrodents (cont.) (83-3(b)) / A NOEL was not established
Statistical problems
Reproduction (83-4) / Information missing from laboratory results
Lack of characterization of the test material
Information missing or requiring clarification of laboratory methods or results
Missing historical controls
A NOEL was not established due to effects at the lowest dose tested
Low fertility and/or inadequate number of animals were used per dose level
A NOEL was not established in the absence of reproductive effects
Metabolism (85-1) / Inadequate or missing data on identification of metabolites
Improper methodology or dosing regimen
Inadequate number of animals were used in the dose groups
No individual animal data
Improper reporting
Inadequate or missing tissue residue analysis data
Testing at only one dose level
Only one sex of animal used
Lack of an intravenous dose group
No collection of 14 CO2
Dermal Penetration (85-2) / Incomplete/missing data evaluation
Improper test material preparation/application
Raw data missing and incomplete summary tables
No signed quality assurance statement
Missing purity or concentration of test material
SOURCE: P. Fenner-Crisp, EPA, personal communication, 1992
The additional acute study proposed in guideline number 81-4 is acute neurotoxicity testing in the rat. This study would be required for all pesticide registrations (food and nonfood) and experimental use permits (EUPs), and it would include assessments of function and activity as well as histopathological (microscopic) examination of selected neural tissue. EPA presently requires that this study be conducted by manufacturers wishing to reregister.
SUBCHRONIC TOXICITY STUDIES
General Description
Subchronic exposures do not elicit effects that have a long latency period (e.g., carcinogenicity). However, they do provide information on health hazards that may result from repeated exposures to a pesticide over a period up to approximately 30% of the lifetime of a rodent. Subchronic tests also provide information necessary to select proper dose levels for chronic studies, especially for carcinogenicity studies for which an MTD must be selected. According to EPA (1984), rats selected for these studies should be started on the test material shortly after weaning, ''ideally before the rats are 6 and, in any case, not more than 8 weeks old." For dogs, dosing should begin when they are 4 to 6 months of age and "not later than 9 months of age."
Most subchronic toxicity studies monitor clinical or behavioral (neurological) signs of toxicity, body weight, food consumption, eye effects, certain plasma or serum and urine parameters, organ weights, and gross and microscopic pathology. Clinical and behavioral signs of toxicity are observed and recorded daily. They can consist of activity, gait, excreta, hair coat, and feeding and drinking patterns. Body weight and food consumption data are routinely recorded throughout the study at intervals (usually weekly) determined by the length of the study. Ophthalmoscopic examinations are conducted at the beginning of the study and, typically, just before it terminates. The laboratory parameters typically examined are summarized in Table 4-5.
The results of hematology testing indicate whether, for example, the chemical affects blood cell formation and survival, clotting factors, and platelets. Clinical chemistry and urinalysis results can indicate possible kidney, liver, pancreas, and cardiac function or toxicity as well as any electrolyte imbalance. Urinalysis results can indicate adequacy of kidney, liver, and pancreas function.
After necropsy, the weights of certain organs are also recorded. These organs generally include brain, gonads, liver, and kidneys, which are the four required according to EPA testing guidelines (EPA, 1984). If toxicity is known to occur in another organ from previous testing, the weight of this organ should also be reported. For thyroid toxicity, for example, the weight of the thyroids should be recorded. Changes from untreated control animals are generally an indication of potential toxicity in this organ.
A complete necropsy is performed after sacrifice or death of the test animal. Generally all tissues are examined, and those saved for microscopic examination are aorta, jejunum, peripheral nerve, eyes, bone marrow, kidneys, cecum, liver, esophagus, colon, lung, ovaries, duodenum, lymph nodes, oviduct, brain, stomach, pancreas, skin, mammary gland, rectum, heart, spleen, spinal cord, testes, musculature, thyroid/parathyroid, pituitary, epididymis, salivary glands, ileum, adrenals, thymus, trachea, urinary bladder, accessory sex organs, and gallbladder.
The data described above [below] are not required for all subchronic studies. For the 21-day dermal study, for example, only limited necropsy data are required.
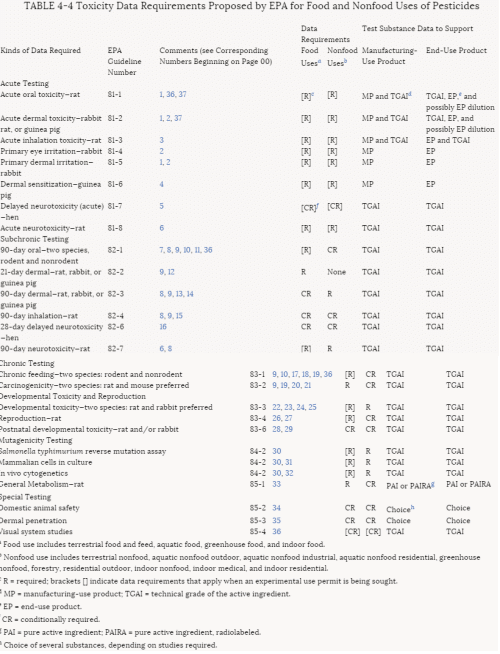
TABLE 4-4 Toxicity Data Requirements Proposed by EPA for Food and Nonfood Uses of Pesticides
a Food use includes terrestrial food and feed, aquatic food, greenhouse food, and indoor food.
b Nonfood use includes terrestrial nonfood, aquatic nonfood outdoor, aquatic nonfood industrial, aquatic nonfood residential, greenhouse nonfood, forestry, residential outdoor, indoor nonfood, indoor medical, and indoor residential.
c R = required; brackets [] indicate data requirements that apply when an experimental use permit is being sought.
d MP = manufacturing-use product; TGAI = technical grade of the active ingredient.
e EP = end-use product.
f CR = conditionally required.
g PAI = pure active ingredient; PAIRA = pure active ingredient, radiolabeled.
h Choice of several substances, depending on studies required.
Notes for Table 4-4: Specific Conditions, Qualifications, or Exceptions to the Designated Test Procedures
1. Not required if test material is a gas or highly volatile.
2. Not required if test material is corrosive to skin or has pH <2 or >11.5; such a product will be classified as toxicity category 1 on the basis of potential eye and dermal irritation effects.
3. Required when the product consists of, or under conditions of use will result in, an inhalable material (e.g., gas, volatile substances, or aerosol/particulate).
4. Required unless repeated dermal exposure does not occur under conditions of use.
5. Required for uncharged organophosphorus esters, thioesters, or anhydrides of organophosphoric, organophosphonic, or organophosphoramidic acids or of related phosphorothioic, phosphonothioic, or phosphorothioamidic acids, or other substances that may cause the neurotoxicity sometimes seen in this class.
6. Additional measurements such as cholinesterase determinations for certain pesticides (e.g., organophosphates and carbamates) may also be required. The route of exposure should correspond to a primary route of human exposure.
7. Required if intended use of the pesticide is expected to result in human exposure via the oral route.
8. All 90-day subchronic studies can be designed to simultaneously fulfill the requirements of the 90-day neurotoxicity study.
9. Studies must include additional end points so as to provide an immunotoxicity screen in the rodent. An equivalent independent study may fulfill the requirements for an immunotoxicity screen.
10. In most cases, where the theoretical maximum residue contribution (TMRC) exceeds 50 percent of the reference dose (Rfd), a 1-year (or longer) interim report on a chronic (2-year) feeding study is required to support a temporary tolerance. This report is to be in addition to the 90-day feeding studies in rodents and nonrodents.
11. If the pesticide is found to leach into groundwater or may contaminate drinking water, a 90-day drinking water study may be required unless data demonstrate that there are no significant differences in toxicity observed when the test material is administered in feed versus when the test material is administered in drinking water. This study may be requested in addition to any 90-day oral studies that may be required.
12. Required if intended use of the pesticide is expected to result in human exposure via the dermal route and data from a subchronic 90-day dermal toxicity study are not required.
13. For nonfood uses, a 90-day dermal toxicity study is required, since intended use of the pesticide is expected to result in repeated dermal exposure of humans.
14. For food uses, required if: (a) the active ingredient of the product is known or expected to be metabolized differently by the dermal route of exposure than by the oral route, and a metabolite of the active ingredient is the toxic moiety; (b) the active ingredient of the product is classified as toxicity category I or II on the basis of acute dermal toxicity data.
15. Required if the active ingredient is a gas at room temperature or if use of the product results in respirable droplets and use may result in repeated inhalation exposure at a concentration likely to be toxic, regardless of whether the major route of exposure is inhalation
16. Required for substances when statistically or biologically significant effects were seen in the acute study (Guideline 81-7), or if other available data indicate that the substance can cause this type of delayed neurotoxicity.
17. Required if either of the following criteria is met: (a) use of the pesticide is likely to result in repeated human exposure over a significant portion of the human life span (e.g., products intended for use in and around residences, swimming pools, and enclosed working spaces or their immediate vicinity); (b) the use requires a tolerance for the pesticide or an exemption from the requirement to obtain a tolerance for the pesticide or an exemption from the requirement to obtain a tolerance, or requires issuance of a food additive regulation.
18. Based on acute and subchronic neurotoxicity studies, and/or on other available data, a functional observational battery, an assessment of motor activity, and perfusion neuropathology may be required.
19. Studies designed to simultaneously fulfill the requirements of both the chronic feeding and carcinogenicity studies (i.e., a combined study) may be conducted. Minimum acceptable study durations for chronic feeding and carcinogenicity studies are as follows: chronic rodent feeding study (food use pesticide)—24 months; chronic rodent feeding study (nonfood pesticide)—12 months in usually sufficient; chronic nonrodent (i.e. dog) feeding study—12 months; mouse carcinogenicity study-18 months; and rat carcinogenicity study—24 months.
20. Required active ingredients or any of their metabolites, degradation products, or impurities are structurally related to a recognized carcinogen, cause mutagenic effects as demonstrated by in vitro or in vivo testing, or produce a morphologic effect in any organ (e.g., hyperplasia, metaplasia) in subchronic studies that may lead to neoplastic change. The use requires a tolerance for the pesticide or exemption from the requirement to obtain a tolerance or requires the issuance of a food additive regulation. Use of the pesticide product is likely to result in exposure of humans over a portion of the life span that is significant in terms of either the timing or duration of exposure (e.g., pesticides used in treated fabrics for wearing apparel, diapers, or bedding; insect repellents applied directly to the skin; swimming pool additives; or constant-release indoor aerosol pesticides).
21. Range-finding studies of at least 90 days duration in rats and mice are generally required to determine dose levels adequate to demonstrate an MTD in carcinogenicity studies. A subchronic 90-day oral study conducted in accordance with Guideline 82-1 may also be acceptable for this purpose.
22. Testing in two species is required for food uses. For products intended for nonfood uses, testing in two species is required if significant exposure of human females of child-bearing age may reasonably be expected. For other nonfood uses, testing in at least one species is required. A study in one species is required to support a temporary tolerance.
23. Testing in a second species is required if significant developmental toxicity is observed after testing in the first species.
24. The test substance or vehicle is usually administered by oral intubation, unless the chemical or physical characteristics of the test substance or pattern of human exposure suggest a more appropriate route of administration.
25. Under certain conditions where a pesticide is determined to be a developmental toxicant (e.g., after oral dosing), additional testing via other routes (e.g., dermal) may be required.
26. Required to support products intended for food and nonfood uses if the use is likely to result in exposure of humans over a portion of the life span that is significant in terms of the frequency, magnitude, or duration of exposure (e.g., pesticides used in treated fabrics for wearing apparel, diapers, or bedding; insect repellents applied directly to the skin; swimming pool additives; or constant-release indoor pesticides used in aerosol form). Also may be required for nonfood uses if adverse effects on organs of the reproductive system are observed in 90-day or other studies, and/or if developmental toxicity is demonstrated by available data (Guideline 83-3).
27. In most cases, where the TMRC exceeds 50% of the RfD, a first-generation (or longer) interim report on a multigeneration reproduction study is required to support a temporary tolerance.
28. Conditionally required to more fully assess any of the manifestations of developmental toxicity. These studies permit assessment of potential functional deficits that cannot be evaluated in the classical developmental toxicity study (Guideline 83-3). Protocols for these studies are usually designed on a case-by-case basis.
29. On the basis of acute and subchronic neurotoxicity studies, and/or on other available data, a developmental neurotoxicity study may be required. For this type of postnatal study, a guideline is available.
30. An initial battery of mutagenicity tests with possible confirmatory testing is minimally required. Also, results from other mutagenicity tests that may have been performed and as complete a reference list as possible shall be submitted. Subsequent testing may or may not be required based on the evidence available to EPA's Office of Pesticide Programs in accordance with the objective and considerations for mutagenicity testing. Current protocols for tests in the initial battery and other mutagenicity tests are given in the EPA's Office of Pesticides and Toxic Substances Health Effects Testing Guidelines (40 CFR Part 798, Subpart F—Genetic Toxicity). Because of the rapid improvements in the field, applicants are encouraged to discuss test selection, protocol design, and results of preliminary testing with the agency.
31. Choice of assays using either mouse lymphoma L5178Y cells, thymidine kinase (tK) gene locus, maximizing assay conditions for small colony expression and detection; Chinese hamster ovary (CHO) or Chinese hamster lung fibroblast (V79) cells, hypoxanthine-guanine phosphoribosyl transferase (hgprt) gene locus, accompanied by an appropriate in vitro test for clastogenicity; or CHO cells strain AS52, xanthine-guanine phosphoribosyl transferase (xprt) gene locus.
32. Choice of assays; initial consideration usually given to rodent bone marrow, using either metaphase analysis (aberrations) or micronucleus assay.
33. Required for all food uses and when chronic and/or carcinogenicity studies are required. Also may be required if significant adverse effects are observed in toxicology studies (e.g., reproduction and developmental toxicity).
34. May be required, on a case-by-case basis, to support registration of an end-use product if cats, dogs, cattle, pigs, sheep, horses, or other domesticated animals will be exposed to the pesticide product, including, but not limited to, exposure through direct application for pest control and consumption of treated feed.
35. Dermal penetration studies are required for compounds that have serious toxic effects, as identified in oral or inhalation studies, and for which a significant route of human exposure is dermal. Thus, this study is required when any of the following exposure studies are required: passive dosimetry—dermal exposure (Guidelines 133-3, 231 or 233), foliar dislodgeable residue dissipation (Guideline 132-1), soil dislodgeable residue dissipation (Guideline 132-1), and indoor surface residue dissipation, unless the toxicity studies (including Guidelines 82-6, 82-7 83-1, 83-2 83-3, 83-4 and 83-6) that triggered the need for these exposure studies were conducted via the dermal route of dosing. Registrants should work closely with the agency in developing an acceptable protocol for performing dermal penetration studies.
36. Special testing (acute, subchronic, and/or chronic) is required for organophosphates, and may be required for other cholinesterase inhibitors and other pesticides that have demonstrated a potential to adversely affect the visual system. Registrants should consult with the agency for development of protocols and methodology prior to initiation of studies.
37. Testing of the end-use product dilution is required if it can be reasonably anticipated that the results of such testing may meet the criteria for restriction to use by certified applicators specified in 40 CFR 152.170(b) or the criteria for initiation of special review specified in 40 CFR 154.7(a)(1).
SOURCE: Code of Federal Regulations, Title 40, Parts 150 to 189, 11992.
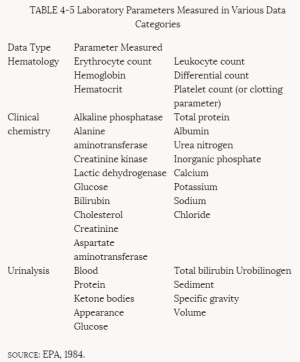
TABLE 4-5 Laboratory Parameters Measured in Various Data Categories
SOURCE: EPA, 1984.