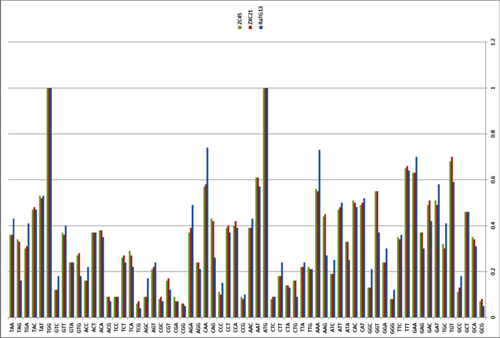
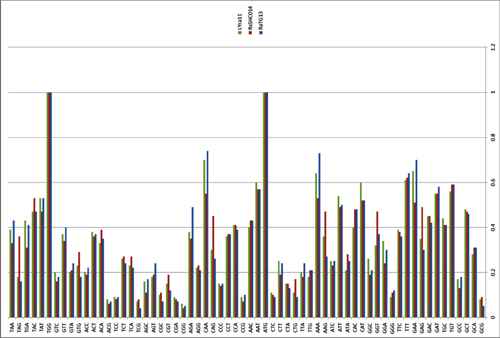
by Lei-Ping Zeng, Yu-Tao Gao, Xing-Yi Ge, Qian Zhang, Cheng Peng, Xing-Lou Yang, Bing Tan, Jing Chen, Aleksei A. Chmura, Peter Daszak, and Zheng-Li Shi
S. Perlman, Editor
University of Iowa
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.
There have been three publications, in 2015,30 2016 and 2017, describing the WIV gain of function research. The WIV, having learned both basic and traceless infectious-clone technology from joint research with a laboratory at the University of North Carolina (UNC) in 2015, initiated construction of novel chimeric coronaviruses without UNC immediately thereafter. WIV’s first publication on the use of basic infectious-clone technology to construct novel chimeric coronaviruses at WIV appeared in 2016.31 WIV’s first publication on the use of traceless, signature-free infectious-clone technology also appeared in 2016.32
-- Did the SARS-CoV-2 virus arise from a bat coronavirus research program in a Chinese laboratory? Very possibly, by Milton Leitenberg
ABSTRACT
Bats harbor severe acute respiratory syndrome (SARS)-like coronaviruses (SL-CoVs) from which the causative agent of the 2002-2003 SARS pandemic is thought to have originated. However, despite the fact that a large number of genetically diverse SL-CoV sequences have been detected in bats, only two strains (named WIV1 and WIV16) have been successfully cultured in vitro. These two strains differ from SARS-CoV only in containing an extra open reading frame (ORF) (named ORFX), between ORF6 and ORF7, which has no homology to any known protein sequences. In this study, we constructed a full-length cDNA clone of SL-CoV WIV1 (rWIV1), an ORFX deletion mutant (rWIV1-ΔX), and a green fluorescent protein (GFP)-expressing mutant (rWIV1-GFP-ΔX). Northern blotting and fluorescence microscopy indicate that ORFX was expressed during WIV1 infection. A virus infection assay showed that rWIV1-ΔX replicated as efficiently as rWIV1 in Vero E6, Calu-3, and HeLa-hACE2 cells. Further study showed that ORFX could inhibit interferon production and activate NF-κB. Our results demonstrate for the first time that the unique ORFX in the WIV1 strain is a functional gene involving modulation of the host immune response but is not essential for in vitro viral replication.
IMPORTANCE
Bats harbor genetically diverse SARS-like coronaviruses (SL-CoVs), and some of them have the potential for interspecies transmission. A unique open reading frame (ORFX) was identified in the genomes of two recently isolated bat SL-CoV strains (WIV1 and -16). It will therefore be critical to clarify whether and how this protein contributes to virulence during viral infection. Here we revealed that the unique ORFX is a functional gene that is involved in the modulation of the host immune response but is not essential for in vitro viral replication. Our results provide important information for further exploration of the ORFX function in the future. Moreover, the reverse genetics system we constructed will be helpful for study of the pathogenesis of this group of viruses and to develop therapeutics for future control of emerging SARS-like infections.
INTRODUCTION
Severe acute respiratory syndrome coronavirus (SARS-CoV) is a zoonotic pathogen that caused the 2002-2003 SARS pandemic, which originated in China (1). Since then, genetically diverse SARS-like coronaviruses (SL-CoVs) have been reported in bats in China, Europe, and Africa (2,–11), indicating a wide geographic distribution of this group of viruses. However, most bat SL-CoVs have been identified only by sequences and are not fully characterized due to the lack of cultured viruses. Thus, their potential for transmission to and likely pathogenesis in domestic animals and humans remain untested. WIV1 and WIV16 are two recently identified SL-CoV strains with high genomic similarity to human SARS-CoV. These two strains have been successfully cultured in vitro and have been shown to use the same molecule (angiotensin-converting enzyme [ACE2]) for cellular entry as SARS-CoV (2, 10). Recently, another bat SL-CoV strain, SHC014, has been demonstrated to use human ACE2 by the construction of an infectious cDNA clone (12). Furthermore, animal infection experiments indicated that SL-CoV WIV1 and SHC014 could replicate efficiently and caused low pathogenesis in ACE2 transgenic mice (12, 13). The fact that the native bat SL-CoVs could use human ACE2 without any mutations indicates a high risk of interspecies transmission for these and similar coronaviruses that may exist in natural reservoirs.
Coronaviruses have the largest genomes among RNA viruses. Their genomes consist of a positive, single-stranded RNA of around 30,000 nucleotides (nt), with two-thirds at the 5′ end encoding genome replication proteins (ORF1ab) and one-third at the 3′ end encoding structural proteins, including a spike glycoprotein (S), a small envelope protein (E), a membrane protein (M), and a nucleocapsid protein (N). Coronaviruses carry a set of open reading frames (ORFs) expressed from full-length mRNAs and subgenomic-length mRNAs (sgRNAs), which have a common 3′ end originating at distinct transcription regulatory sequences (TRS) and joined with a common leader sequence encoded at the 5′ end of genomic RNA (14). Currently, coronaviruses are divided into the genera Alphacoronavirus, Betacoronavirus, and Gammacoronavirus and the proposed genus Deltacoronavirus (15). SARS-CoV and SL-CoVs are grouped into the same coronavirus species, SARS-related coronavirus (SARSr-CoV), within the genus Betacoronavirus. Besides the family-conserved genes, SARSr-CoV possesses several accessory genes, including ORF3, ORF6, ORF7, ORF8, and ORF9, which are specific for this group of coronaviruses but not essential for in vitro viral replication (16,–18). Accessory genes in coronavirus genomes play important roles in regulating the host immune response (19). The SARS-CoV ORF3a, ORF3b, and ORF6 have been reported to inhibit the host interferon (IFN) response during virus infection and contribute to pathogenesis (20, 21). ORF3a and ORF7a activate NF-κB and upregulate interleukin-8 (IL-8) and CCL5 production (22, 23). Bat SL-CoVs display great genetic diversity and share overall nucleotide sequence identities of 88 to 97% with human SARS-CoV (2,–11). Bat SL-CoVs WIV1 and WIV16 are the closest relatives to human SARS-CoV discovered so far. These two viruses are identical in genomic structures except that WIV1 and -16 have an extra ORF (named ORFX) between ORF6 and ORF7 with no homology to any known protein sequences (2, 10).
In this study, we explored the function of ORFX in modulating the host immune response through the use of eukaryotic overexpression assays and recombinant viruses generated through reverse genetics techniques.
MATERIALS AND METHODS
Virus and cells.
The SL-CoV WIV1 strain (GenBank accession number KF367457) and other viruses were propagated as described previously (2). Sendai virus (SeV) strain Cantell (kindly provided by Hanzhong Wang) was propagated in 10-day-old embryonated chicken eggs at 37°C for 48 h (24). All experiments using live virus was conducted under biosafety level 2 (BSL2) conditions. HeLa cells stably expressing human ACE2 (HeLa-hACE2) were described previously (25). 293T, Vero E6, HeLa, and HeLa-hACE2 cells were grown and propagated in Dulbecco's modified Eagle's medium (GIBCO, Invitrogen) supplemented with 10% fetal bovine serum (Life Technologies). Calu-3 cells were grown and propagated in Dulbecco's modified Eagle's medium–nutrient mixture F-12 medium supplemented with 15% fetal bovine serum. Cells were grown at 37°C in a humidified atmosphere with 5% CO2.
Plasmids.
The coding region of ORFX was amplified by reverse transcription-PCR (RT-PCR) from viral RNA using the Superscript one-step RT-PCR kit (Invitrogen). The amplified gene was cloned into plasmid pCAGGS with a C-terminal hemagglutinin (HA) tag (pCAGGS-ORFX) for eukaryotic expression. Reporter plasmids used included pIFNκ-Luc (expressing firefly luciferase under the control of the IFN-β promoter), pNF-κB-Luc (expressing firefly luciferase under the control of the NF-κB promoter), and pRL-TK (expressing Renilla luciferase under the control of the herpes simplex virus thymidine kinase promoter), as well as an expression plasmid for influenza virus NS1, as described previously (24). Plasmids expressing subcellular organelle markers, including SecG1β-green fluorescent protein (GFP) (endoplasmic reticulum [ER] marker), B4Gal-Ti-red fluorescent protein (RFP) (Golgi apparatus marker), and Mito-yellow fluorescent protein (YFP) (mitochondrion marker), were kindly provided by Yanyi Wang of the Wuhan Institute of Virology.
Viral infection assays.
Vero E6, Calu-3, and HeLa-hACE2 cells were infected with viruses at a multiplicity of infection (MOI) of 1.0, 0.1, or 0.001 in 25-cm2 flasks with a 1-h adsorption period, followed by two washes with D-Hanks solution and culturing by adding 3 ml of medium. The viral supernatants were harvested, at 0, 2, 6, 12, 18, 24, 36, 48, and 72 h postinoculation, with 300 μl removed and 300 μl medium added back at each time point. The virus concentration was titrated by plaque assay in Vero E6 cells.
Vero E6 cells were infected by rWIV1-GFP-ΔX or mock infected. After 24 h, fluorescence micrographs was taken to check the expression of green fluorescent protein.
Cloning of WIV1 cDNAs.
The virus genome was divided into 8 continuous fragments (A to G) and amplified using specific primers (primer sequences are available upon request). Viral RNA was extracted from the supernatant of WIV1-infected cultures and reverse transcribed with Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega) and random hexamer deoxynucleotide primers. The cDNA was denatured for 5 min at 95°C and amplified by PCR with KOD DNA polymerase (Toyobo) for 20 cycles of 95°C for 30 s, 60°C for 30 s with a 0.5°C decrease per cycle, and 68°C for 5 min, 15 cycles of 95°C for 30 s, 50°C for 30 s, and 68°C for 5 min, and a final extension at 68°C for 10 min. The amplicons were cloned into pGEM-T Easy (Promega). Besides three natural BglI sites, several BglI sites were introduced by synonymous mutations in the PCR process to make all contiguous cDNA fragments capable of unidirectional ligation. SacII and AscI sites were introduced into the 5′ terminus of fragment A and the 3′ terminus of fragment G, respectively. A poly(A) sequence (25 nt) was added to the 3′ terminus of fragment G. At least three colonies of each cDNA clone were sequenced, and the one identical to or with some synonymous mutations to the reported sequence was selected for assembly.
To ablate a natural BglI site at position 1575, primers FA, F-c1575a, R-c1575a, and RA were used for overlap extension PCR (OE-PCR) to introduce the synonymous mutation C1575A (primer sequences are available upon request). Based on previous in vitro transcription tests, the synonymous mutation T27527C was also introduced to interrupt a potential T7 termination site via OE-PCR.
Strategy for modifying pBeloBAC11.
The cytomegalovirus (CMV) promoter was amplified from pcDNA3.1(+) (Thermo Fisher Scientific) with forward primer 5′-TGAGGATCCCGTTGACATTGATTATTGACTAG-3′ and reverse primer 5′-CCTGACTGCAGGTCGACTGCCGCGGAGCTCTGCTTATATAGACC-3′. Hepatitis delta virus (HDV) ribozyme was synthesized as described previously (26), and amplified with forward primer 5′-CAGTCGACCTGCAGTCAGGCGCGCCGGGTCGGCATGGCATCTCC-3′ and reverse primer 5′-CTAGAAGGCACAGCTCCCTTAGCCATCCGAGTGG-3′. The bovine growth hormone (BGH) transcription terminal signal was amplified from pcDNA3.1(+) with forward primer 5′-GGATGGCTAAGGGAGCTGTGCCTTCTAGTTGCCAGC-3′ and reverse primer 5′-TGAAAGCTTCCATAGAGCCCACCGCATCC-3′. The three PCR products then were ligated using OE-PCR, with BamHI and HindIII sites flanking the amplicon and SacII and AscI sites between the CMV promoter and HDV ribozyme. The amplicon was then inserted into pBeloBAC11 (New England BioLabs) between BamHI and HindIII sites. The construct was designated pBAC-CMV.
Construction of infectious bacterial artificial chromosome (BAC) clones of WIV1.
Subclone A and subclone G were first digested with SacII and AscI (New England BioLabs), respectively, followed by treatment with calf intestinal alkaline phosphatase (CIAP) (TaKaRa), chloroform extraction, and isopropanol precipitation, and then restricted with BglI (TaKaRa). Subclones B to F were digested with BglI. pBAC-CMV was digested with SacII and AscI. All digestion products were then separated using 1% agarose gels, excised, and purified by using a gel extraction kit (Omega). Digested fragments A to G and pBAC-CMV were ligated overnight at 4°C, transformed into DH10B competent cells, and plated on Chl+ LB culture. Ten clones were screened by restriction fragment length polymorphism (RFLP) analysis with NcoI, StuI, or HindIII. The correct clone was named pBAC-CMV-rWIV1 (Fig. 1).
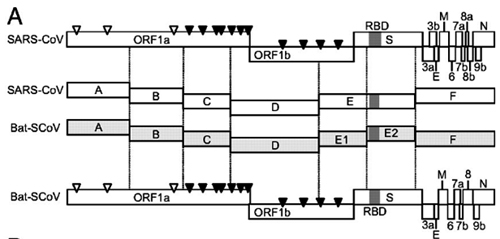
FIG 1. Strategy for construction of an infectious WIV1 BAC clone. (A) Genomic structure of WIV1. (B) The mutations are indicated under the stars. C1575A was used to ablate a natural BglI site at nucleotide 1571 (▽), and T27527C was used to disrupt a potential T7 stop site. The others were for introducing BglI sites (▼). (C) The WIV1 genome was split into eight contiguous cDNAs (A to G): A, nt 1 to 4387; B, nt 4388 to 8032; C1, nt 8033 to 10561; C2, nt 10562 to 12079; D, nt 12080 to 17017; E, nt 17018 to 22468; F, nt 22469 to 27352; G, nt 27353 to 30309. Unique BglI sites were introduced into the fragments by synonymous mutations to make these fragments capable of unidirectional ligation along with native BglI sites in the genome. The original nucleotides are shown above the flanking sequences of corresponding fragments. A poly(A) sequence was added to the 3′ terminus of fragment G. A CMV promoter, HDV ribozyme, and BGH transcriptional terminal signal were inserted into pBeloBAC11 between BamHI and HindIII sites. SacII and AscI sites were introduced between the CMV promoter and ribozyme. Fragments A to G were inserted into the pBAC-CMV plasmid in a single step.
Construction of WIV1 mutants.
To delete ORFX, the fragment F was PCR amplified with primers FF (5′-ACCTGTGCCCTTTTGGCGAGGTTTTTAATGCTACTAC-3′) and RFox (5′-GCCTCTAGGGCTCAAGGATAATCTATCTCCATAGG-3′). Fragment G was PCR amplified with FGox (5′-GCCCTAGAGGCAACGAACATGAAAATTATTCTCTTCC-3′) and RG (5′-ACTGGCGCGCCTTTTTTTTTTTTTTTTTTTTTTTTTGTCATTCTCCTGAGAAGC-3′). This new fragment was named Gox. These two products were then cloned into pGEM-T Easy. The two fragments were inserted into the BAC along with the other fragments as described above. The rescued mutant was named as rWIV1-ΔX. To place GFP into the open reading frame of ORFX, the F fragment was amplified with primers FF and RFoeGFP (5′-GCTCACCATAGTGGTTCGTTTATCAAGGATAATCTATCTCC-3′). The GFP gene was amplified with primers 5′-CCTTGATAAACGAACCACTATGGTGAGCAAGGGCGAGGAGC-3′ and 5′-TGCCTCTAGGGCTTACTTGTACAGCTCGTCCATGCC-3′. The two PCR products were ligated by OE-PCR, and the product was inserted into pGEM-T Easy. The rescued mutant was named rWIV1-GFP-ΔX.
Transfection of infectious WIV1 BAC clones.
Vero E6 cells were seeded in a 6-well plate a day in advance, and then one well was transfected with 6 μg infectious BAC plasmids constructed as described above with Lipofectamine LTX and Plus reagent (Life Technologies). Virus progeny was plaque purified once. One clone was passaged once in Vero E6 cells for 72 h and used to generate a stock for future use.
RFLP.
RNAs extracted from wild-type and recombinant viruses were reverse transcribed with random hexamer primers. RT-PCR was used to generate five amplicons containing the five mutations designed in the strategy. These amplicons included a 1,124-bp amplicon (nucleotide positions 1312 to 2435) spanning a naturally occurring BglI site at nucleotide 1571 that had been ablated in recombinant viruses, a 1,438-bp amplicon spanning the B/C1 junction (nucleotide positions 7560 to 8997), a 1,437-bp amplicon spanning the C1/C2 junction (nucleotide positions 10196 to 11632), a 1,437-bp amplicon spanning the D/E junction (nucleotide positions 16793 to 18229), and a 1,438-bp amplicon spanning the E/F junction (nucleotide positions 21908 to 23345) (these amplicons correspond to fragments F1 to F5 in Fig. 1). The first amplicon of wild-type WIV1 (wtWIV1) that contains nucleotide 1571 can be cleaved by BglI, but the other four amplicons cannot. In contrast, the five amplicons of recombinant viruses are different from those of wild-type virus in the capability of being cut by BglI.
Northern blot analysis.
The N gene was amplified with primers WIV1-NF (5′-ATGTCTGATAATGGACCCCA-3′) and WIV1-3R (5′-GTCATTCTCCTGAGAAGCTA-3′) and used as a template for probe preparation according to the description in the DIG-High Prime DNA labeling and detection starter kit II (Roche). Vero E6 cells were infected with wild-type and recombinant viruses at an MOI of 1.0. At 24 h postinfection, intracellular RNA was isolated using TRIzol reagent (Ambion). RNA (20 μg) was precipitated, treated with 17 μl sample buffer (50% formamide, 2.2 M formaldehyde [37%], 1× morpholinepropanesulfonic acid [MOPS]) at 65°C for 10 min, supplemented with 3 μl 10× dye solution (50% glycerol, 0.25% bromophenol blue, 0.25% xylene cyanole FF), and then separated in a denaturing 0.8% agarose–2.2 M formaldehyde gel at 28 V for ∼17 h. The RNA was hydrolyzed with 0.05 M NaOH for 40 min, transferred to a Hybond-N+ membrane (GE Healthcare) for ∼18 h, and then cross-linked to the membrane using UV light. The membrane was prehybridized, probed with a digoxigenin (DIG)-labeled probe for the N gene, and washed, and detection was performed according to the instructions for the DIG-High Prime DNA labeling and detection starter kit II (Roche).
RT-PCR of leader-containing transcripts.
Intracellular RNA was isolated from wtWIV1. A forward primer (Leader-F) located in the leader sequence, along with various reverse primers located in several ORFs, was used for amplifying leader-containing sequences (primer sequences are available upon request). Leader-containing amplicons were sequenced with the corresponding reverse primers.
ORFX subcellular location.
HeLa cells were transfected with an ORFX-expressing plasmid and cotransfected with organelle markers expressing plasmid SecG1β-GFP, B4Gal-Ti-RFP, or Mito-YFP. After 24 h, the cells were fixed and stained with a mouse anti-HA IgG (Promoter). A Cy3-conjugated goat anti-mouse IgG (Promoter) was used for secondary detection in cells expressing ER or mitochondrial markers. A fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Promoter) was used for secondary detection in cells expressing the Golgi marker. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Staining patterns were examined with an Olympus Fluoview upright confocal microscope (Olympus).
Luciferase assays and quantitative PCR.
For the ORFX-mediated IFN promoter assay, 293T cells were seeded in 12-well plates and cotransfected with empty vector plasmid pCAGGS, plasmid pCAGGS-NS1, or increasing amounts (100, 200, 400, 600, and 800 ng) of pCAGGS-ORFX with the indicated reporter plasmids. At 24 h posttransfection, cells were infected with Sendai virus (SeV) (100 hemagglutinin units [HAU]/ml) for 12 h to induce IFN production or were treated with tumor necrosis factor alpha (TNF-α) for 1 h to activate NF-κB. Cell lysates were prepared, and luciferase activity was measured using dual-luciferase assay kits (Promega) according to the manufacturer's instructions.
293T cells were transfected with empty vector, NS1-expressing plasmid, or increasing amounts (100, 300, and 600 ng) of ORFX-expressing plasmid. After 24 h, the cells were infected with SeV (100 HAU/ml). At 12 h postinfection, the cells were lysed. The mRNA was extracted and reverse transcribed with PrimeScript RT master mix (TaKaRa). The expression level of IFN-β mRNA was determined by quantitative PCR using SYBR Premix Ex Taq II (TaKaRa). The GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was quantified as an inner control. 293T cells were transfected as described above. After 24 h, the cells were treated with TNF-α for 6 h, and the cell RNA was extracted and used for quantification of the expression of IL-8 mRNA. All experiments were performed in triplicate and repeated at least three times. All primer sequences used in the quantitative PCRs are available upon request.
IRF3 translocation assay.
293T cells were transfected with empty vector, NS1, or ORFX-expressing plasmid. After 24 h, IFN regulatory factor 3 (IRF3) nuclear translocation was induced by infecting the cells with SeV for 8 h. The cells were fixed and stained with a rabbit anti-IRF3 polyclonal IgG (Proteintech) and a mouse anti-HA IgG (Promoter). An Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Yeasen) and an Alexa Fluor 555-conjugated donkey anti-mouse IgG (Beyotime) were used to detect IRF3 and ORFX, respectively. The cells transfected with empty vector were stained with a rabbit anti-IRF3 polyclonal IgG and a goat anti-SeV IgG (kindly provided by Lin-Fa Wang, Duke-NUS Graduate Medical School, Singapore) as an indication of infection efficiency. An Alexa Fluor 488-conjugated donkey anti-rabbit IgG and a Cy3-conjugated donkey anti-goat IgG (Promoter) were used to detect IRF3 and SeV, respectively. Nuclei were stained with DAPI.
Quantification of mRNA expression of cytokines in infected Calu-3 cells.
Calu-3 cells grown in 24-well plates were mock infected or infected with rWIV1 or rWIV1-ΔX at an MOI of 5 or with SeV (100 HAU/ml). The cells were lysed at 4, 12, 24, and 30 h postinfection. The mRNA expression levels of IFN-β, IL-6, IL-8, and TNF-α were quantified by quantitative PCRs. The expression of GAPDH mRNA was measured as an internal control. All primer sequences used in the quantitative PCRs are available upon request. The experiment was performed twice.
IFN-β sensitivity assay.
Vero E6 cells were seeded a day in advance. The cells were pretreated with 10, 100, or 1,000 U/ml IFN-β (PBL, Piscataway, NJ) for 24 h, infected with wtWIV1, rWIV1, and rWIV1-ΔX at an MOI of 0.1 PFU/cell, and posttreated with the same amount of IFN-β as used previously. At 24 h postinfection, the viral replication was analyzed by plaque assay. The experiment was performed in triplicate.
Statistics.
The statistical significance of the obtained data was analyzed using a Student t test in GraphPad Prism (GraphPad Software, San Diego, CA). A P value of <0.05 was considered statistically significant. Data are presented as the means ± standard errors of the means (SEM).
RESULTS
Strategy for construction of an infectious WIV1 BAC.
Originally, the genome was split into seven contiguous cDNAs (A to G) (Fig. 1A and andC).C). Due to plasmid instability, fragment C was separated into two segments (C1 and C2). Besides three naturally occurring BglI sites (GCCNNNN↓NGGC), four BglI sites were successfully introduced by synonymous mutations in the genome (Fig. 1B). Different asymmetric 3-nt overhangs at the junctions of each two contiguous fragments were created by these BglI sites. The eight fragments were then linked in one direction. A SacII site was added to the 5′ terminus of fragment A. A poly(A) sequence (25 nt) and an AscI site were added to the 3′ terminus of fragment G. A naturally occurring BglI site at nucleotide 1571 was removed by the synonymous mutation C1575A (Fig. 1B). Other unexpected synonymous mutations also occurred, including T1422C, T12984C, T14213C, T17130C, C17934T, and T26068G.
The plasmid pBAC-CMV was constructed by inserting the cytomegalovirus (CMV) promoter, hepatitis delta virus (HDV) ribozyme, and bovine growth hormone (BGH) transcription terminal signal sequences into pBeloBAC11, along with the introduction of the SacII and AscI sites between the CMV promoter and HDV ribozyme (Fig. 1C). The eight genomic fragments were inserted into pBAC-CMV in one step. Recombinant viruses could be rescued by direct transfection with the BAC constructs.
Rescue of recombinant viruses.
To rescue recombinant WIV1 (rWIV1), fragments A and G were digested with SacII and AscI, respectively. Following calf intestinal alkaline phosphatase (CIAP) dephosphorylation, the two fragments, along with fragments B to F, were digested using BglI and inserted into pBAC-CMV between SacII and AscI sites in one step. The constructed clone (pBAC-CMV-rWIV1) was transfected into Vero E6 cells. A cytopathic effect was observed at 72 h posttransfection. The one ablated natural BglI site and four introduced BglI sites in the rescued viral genome were confirmed by restriction fragment length polymorphism (RFLP) analysis with BglI digestion (Fig. 2A). Using this method, we also rescued an ORFX deletion mutant virus (rWIV1-ΔX) (Fig. 2B, lane 2) and a mutant with a GFP sequence placed in the coding region of ORFX (rWIV1-GFP-ΔX) (Fig. 3A).
FIG 2. Recovery and characterization of recombinant viruses. (A) Restriction fragment length polymorphism. Amplicons flanking five mutated sites of wild-type and recombinant viruses were digested by BglI. The first amplicon (F1) of wild-type virus can be digested by BglI, and its other four amplicons (F2 to F5) cannot be. In contrast, for amplicons of rWIV1, the first amplicon (F1) cannot be digested by BglI and its other four amplicons (F2 to F5) can be. Lane M, DL2000 DNA ladder (TaKaRa). (B) Detection of viral genomic transcription and replication by Northern blotting. Vero E6 cells were infected with wild-type or recombinant viruses, and intracellular RNA was extracted for Northern blot analysis. Lane 1, wtWIV1; lane 2, rWIV1-ΔX; lane 3, rWIV1; lane 4, uninfected control. (C) Growth kinetics of wild-type and recombinant viruses. Vero E6 cells were infected with wtWIV1 (■), rWIV1 (♢), or rWIV1-ΔX (▲) at an MOI of 1.0 or 0.1 PFU/cell. Cell supernatants were taken at the indicated time points postinfection, and virus titers were determined by plaque assay in Vero E6 cells.
FIG 3. Expression and subcellular location of ORFX protein. (A) The open reading frame of ORFX was replaced by the GFP sequence, and the recombinant virus was rescued. Vero E6 cells were infected with the recombinant virus or mock infected. Green fluorescence was visualized at 24 h postinfection. (B) ORFX protein with an HA tag at the C terminus was expressed in HeLa cells, along with SecG1β-GFP (ER marker), Mito-YFP (mitochondria marker), or B4Gal-Ti-RFP (Golgi marker). The cells were fixed after 24 h and stained with a mouse anti-HA IgG. A Cy3-conjugated goat anti-mouse IgG was used for secondary detection in cells expressing an ER or mitochondrial marker. An FITC-conjugated goat anti-mouse IgG was used for secondary detection in cells expressing a Golgi marker. ORFX protein showed a cytoplasmic distribution and colocalized with the ER maker SecG1β.
ORFX is a functional gene not essential for virus replication.
The one-step growth curves for the two rescued recombinant viruses (rWIV1-ΔX and rWIV1) and wild-type WIV1 (wtWIV1) determined by plaque assay showed that rWIV1-ΔX and rWIV1 both replicated to titers close to those of wild-type virus (Fig. 2C). The expected set of appropriately sized 10 sgRNAs, including sgRNA7 (ORFX), were observed in Northern blot analysis in cells infected with wtWIV1 and rWIV1 (Fig. 2B, lanes 1 and 3). As expected, sgRNA7 was not observed in rWIV1-ΔX infected cells (Fig. 2B, lane 2). Analysis of leader-containing sequences indicated that all 10 sgRNAs in wtWIV1 share an identical core sequence, ACGAAC (Table 1), which further confirmed that ORFX is expressed as sgRNA7. The fact that GFP was expressed in rWIV1-GFP-ΔX-infected cells further confirmed that the open reading frame of ORFX could be expressed (Fig. 3A). Subcellular location analyses showed that the ORFX protein colocalized with the ER marker but not with the Golgi and mitochondrial markers (Fig. 3B).
TABLE 1: Leader-containing sequences of sgRNAs
sgRNA / ORF(s) / Leader-containing sequencea / Consensus sequence positions
1 / 1a/b / GTAGATCTGTTCTCTAAACGAACTTTAAAATCTGT / 67–72
2 / S / GTAGATCTGTTCTCTAAACGAACATGAAATTGTTA / 21486–21491
3 / 3a/b / GTAGATCTGTTCTCTAAACGAACTTATGGATTTGT / 25263–25268
4 / E / GTAGATCTGTTCTCTAAACGAACTTATGTACTCAT / 26112–26117
5 / M / GTAGATCTGTTCTCTAAACGAACTAACTATTATTA / 26351–26356
6 / 6 / GTAGATCTGTTCTCTAAACGAACGCTTTCTTATTA / 26916–26921
7 / X / GTAGATCTGTTCTCTAAACGAACCACTATGTTACT / 27272–27277
8 / 7a/b / GTAGATCTGTTCTCTAAACGAACATGAAAATTATT / 27794–27799
9 / 8 / GTAGATCTGTTCTCTAAACGAACATGAAACTTCTC / 28300–28305
10 / N / GTAGATCTGTTCTCTAAACGAACAAACTAAAATGT / 28672–28677
aThe consensus sequence is in bold. Underlining indicates the initiation codon.
ORFX protein inhibits production of IFN-β.
To determine whether ORFX inhibits the induction of IFN, 293T cells were transfected with plasmids pIFNβ-Luc and pRL-TK and a plasmid expressing ORFX, influenza virus strain PR8 NS1 (positive control), or empty vector (negative control). As expected, SeV activated IFN production in cells transfected with empty vector. The positive control, influenza virus NS1 protein dramatically inhibited the expression from the IFN promoter. ORFX protein exhibited an inhibition effect, but the effect decreased when more ORFX protein was expressed (Fig. 4A). Similar results were observed for IFN-β mRNA quantification (Fig. 4B and andCC).
FIG 4. ORFX protein inhibits the production of type I interferon. (A and B) 293T cells seeded in 12-well plates were transfected with 100 ng pIFN-β-Luc, 5 ng pRL-TK, empty vector, an influenza A NS1-expressing plasmid, or increasing doses (100, 200, 400, 600, and 800 ng) of an ORFX-expressing plasmid. Empty vector was added appropriately to ensure that cells in each well were transfected with the same amount of plasmids. The cells were infected with Sendai virus (100 hemagglutinating units/ml) at 24 h posttransfection. Samples were collected at 12 h postinfection, followed by dual-luciferase assay. The results were expressed as the firefly luciferase value normalized to that of Renilla luciferase. The relative expression of IFN-β mRNA was determined by quantitative RT-PCR and normalized to the expression level of GAPDH mRNA. (C) The expression of the NS1 and ORFX proteins was analyzed by Western blotting with an antibody against HA tag. The experiments were replicated three times. (D and E) For the IRF3 translocation assay, 293T cells were transfected with empty vector-, NS1-, or ORFX-expressing plasmid. After 24 h, the cells were infected with Sendai virus to induce IRF3 nuclear translocation. The cells were fixed at 8 h postinfection and stained with anti-HA IgG. A goat anti-Sendai virus polyclonal IgG was used to stain the cells transfected with empty vector. A rabbit anti-IRF3 polyclonal IgG was used to label IRF3. The white arrow indicates IRF3 nuclear translocation. The relative IRF3 translocation ratios were calculated for each group by counting the number of IRF3 nuclear translocation cells (randomly selected from at least 4 fields) and dividing by the total number of infected or transfected cells. The IRF3 nuclear translocation efficiency of each group was expressed as the percentage of their relative IRF3 translocation ratios to that of the control (cells transfected with empty vector). (F) Calu-3 cells were mock infected or infected with rWIV1 or rWIV1-ΔX (MOI of 5) or SeV (100 HAU/ml). At 4, 12, 24, and 30 h postinfection, the cell RNA was extracted and used for quantitative RT-PCR of the expression level of IFN-β mRNA. The experiment was performed in triplicate and replicated twice. (G) Vero E6 cells were pretreated with indicated amount of IFN-β, infected with wtWIV1, rWIV1, or rWIV1-ΔX at an MOI of 0.1 PFU/cell, and posttreated with IFN-β. Viral replication was analyzed at 24 h postinfection by plaque assay. The experiment was performed in triplicate and replicated twice. The differences between selected groups were significant, with P values of less than 0.05, as follows: 0.0049 (*; bars 4 and 6 in panel A), 0.0008 (**; bars 6 and 7 in panel A), 0.0072 (*; bars 4 and 6 in panel B), 0.018 (*; bars for rWIV1 and rWIV1-ΔX in panel F), and <0.0001 (*** in panel G).
An IRF3 nuclear translocation assay was performed to see whether ORFX protein inhibits IFN production through inhibiting this process. 293T cells were transfected with an empty vector-, NS1-, or ORFX-expressing plasmid. After 24 h, IRF3 nuclear translocation was induced by infection with SeV for 8 h. The relative IRF3 translocation ratios were calculated for each group by counting the number of the IRF3 nuclear translocation cells (randomly selected from at least 4 fields) divided by the number of total infected or transfected cells. The IRF3 nuclear translocation efficiency of each group was expressed as the percentage of their relative IRF3 translocation ratios to that of the control (cells transfected with empty vector). As expected, NS1 strongly inhibited translocation of IRF3, while ORFX protein also showed inhibition of IRF3 translocation but less efficiently (Fig. 4D and andEE).
To further investigate the IFN inhibition activity of ORFX, the deletion mutant and wild-type recombinant virus were used to infect Calu-3 cells at an MOI of 5. Mock-infected cells were used as negative control. Calu-3 cells infected with SeV were used as positive control. Samples were collected at 4, 12, 24, and 30 h postinfection. The relative expression of IFN-β mRNA was determined by quantitative PCR and normalized to the expression of GAPDH mRNA. Compared to SeV, WIV1 recombinants induced low levels of IFN-β mRNA in Calu-3 cells (Fig. 4F). The ORFX deletion mutant induced a significantly higher level of IFN-β mRNA than wild-type recombinant virus in infected cells at 12 h postinfection, but there were no significant differences at 24 and 30 h postinfection (Fig. 4F). These results indicate that ORFX protein may play a role in antagonizing IFN only at early times during WIV1 infection.
An ORFX deletion mutant shows increased sensitivity to IFN-β.
To further investigate the effect of ORFX on the viral sensitivity of IFN, we tested the replication efficiencies of wtWIV1, rWIV1, and rWIV1-ΔX in Vero E6 cells which were pretreated and posttreated with IFN-β. The replication of rWIV1-ΔX was inhibited and reduced by ∼0.5 log compared to that of wtWIV1 and rWIV1 at concentrations of 10 and 100 U/ml IFN-β (Fig. 4G), whereas at a higher IFN-β concentration (1,000 U/ml), the rWIV1-ΔX titers did not show an obvious decrease compared to those of wild-type virus. We expected that the ORFX deletion mutant would replicate less efficiently than the wild-type virus in IFN-competent cells. However, we did not find a significant difference when we grew the two viruses in Calu-3 and HeLa-hACE2 cells, even at a very low MOI of 0.001 (Fig. 5).
FIG 5. Comparison of viral replication efficiencies of rWIV1-ΔX and rWIV1 in IFN-competent cells. Calu-3 (A) and HeLa-hACE2 (B) cells were infected with rWIV1 or rWIV1-ΔX at an MOI of 0.001. Samples were collected at 0, 12, 24, 36, 48, 72, 96, and 120 h postinfection. The viral titers were measured by plaque assay.
ORFX protein activates NF-κB.
NF-κB plays an important role in regulating the immune response to viral infection and is also a key factor frequently targeted by viruses for taking over the host cell (27). Several proteins (Nsp1, N, and 3a) encoded by SARS-CoV have activities in both IFN antagonism and NF-κB activation (28). In this study, we also tested whether ORFX protein could activate NF-κB. 293T cells were transfected with pNF-κB-Luc, pRL-TK, empty vector, NS1, or increasing amounts (200, 400, and 600 ng) of ORFX expressing plasmid. After 24 h, the cells were mock treated or treated with TNF-α for 6 h, and luciferase activity was determined. ORFX protein obviously activated NF-κB, no matter whether the cells were treated with TNF-α or not (Fig. 6A), whereas IL-8 was upregulated only when the cells were treated with TNF-α (Fig. 6B). However, no significant difference was observed for IL-6 and IL-8 transcription levels between the rWIV1-ΔX- and rWIV1-infected Calu-3 cells (Fig. 6C and andD).D). A significant difference was observed only for the induction of TNF-α mRNA at the late time of virus infection, when the ORFX deletion mutant induced less TNF-α mRNA (Fig. 6E).
FIG 6. ORFX protein activates NF-κB. 293T cells were transfected with 100 ng pNF-κB-Luc, 10 ng pRL-TK, empty vector, an NS1-expressing plasmid, or increasing amounts (200, 400, and 600 ng) of an ORFX-expressing plasmid. After 24 h, the cells were treated with TNF-α. (A) Dual-luciferase activity was determined after 6 h. The results were expressed as the firefly luciferase activity normalized to that of Renilla luciferase. (B) The relative expression of IL-8 mRNA was quantified through quantitative RT-PCR and normalized to that of GAPDH mRNA. Differences between selected groups were significant, with P value less than 0.05, as follows: <0.0001 (***; bars 1 and 3 in panel A), 0.0339 (*; bars 4 and 7 in panel A), and 0.0002 (***; bars 4 and 6 in panel B). n.s., not significant. The experiments were performed three times. (C to E) The RNA extracted from Calu-3 cells for Fig. 4 was used for quantification of the expression of IL-6 (C), IL-8 (D), and TNF-α (E) mRNAs.
DISCUSSION
In this study, we have developed a fast and cost-effective method for reverse genetics of coronaviruses by combining two approaches developed by others (29, 30). Our method allows the genomes of coronaviruses to be split into multiple fragments and inserted into a BAC plasmid with a single step. Recombinant viruses can then be efficiently rescued by direct transfection of the BAC constructs. As the genomes can be divided into multiple short fragments, mutations can be introduced into individual fragments easily (31). Using this method, we successfully rescued three recombinant viruses derived from SL-CoV WIV1 (rWIV1, rWIV1-ΔX, and rWIV1-GFP-ΔX). The recombinant rWIV1 and rWIV1-ΔX replicated to titers close to those of wtWIV1 in Vero E6 cells (Fig. 2C), suggesting that the deletion of ORFX did not affect WIV1 replication in vitro. Northern blotting and fluorescence microscopy further confirmed that ORFX is transcribed as sgRNA7 and translated in virus-infected cells. These results demonstrated that the unique ORFX in SL-CoV WIV1 is a functional gene but is not essential for virus replication. We propose that the ORFX sgRNA is the template for the translation of a novel 11-kDa accessory protein of WIV1, bringing the total number of group-specific accessory proteins to ten.
In previous studies, it has been proved that SARS-CoV group-specific accessory genes ORF3b and ORF6 inhibit host IFN production and/or signaling during virus infection and contribute to viral pathogenesis (20). It is interesting to know whether the ORFX has a similar function in antagonizing IFN. In this study, ORFX protein showed an inhibitory effect on IFN production, but the effect decreased when more ORFX protein was expressed (Fig. 4A and andB).B). Moreover, the ORFX deletion mutant had a significantly lower inhibitory effect on IFN production than wild-type recombinant virus in infected Calu-3 cells, but only at an early time after infection (Fig. 4F). Furthermore, the IFN sensitivity assay indicated that the ORFX deletion mutant was more sensitive to IFN-β (Fig. 4G), suggesting that ORFX protein may participate in subverting the antiviral state stimulated by IFN-β. All these results suggested that ORFX participates in the modulation of the IFN response. Previous studies showed that SARS-CoV ORF3a and ORF7a activate NF-κB and upregulate IL-8 and CCL5 production (22, 23). In our study, we also found through a dual-luciferase assay that overexpressed ORFX can activate NF-κB (Fig. 6A). Furthermore, the level of TNF-α mRNA induced by wild-type recombinant virus was significantly higher than that induced by the ORFX deletion mutant, but only at the late stage of infection (Fig. 6E). These results indicated that ORFX also participates in activation of NF-κB. We noted that the IFN inhibition activity of ORFX was not dose dependent and decreased when there was more ORFX expression. One possible hypothesis is that ORFX inhibits IFN only at the early stage of infection. At the late stage, it activates NF-κB, which in turn stimulate IFN expression, and this leads to the attenuation of its IFN antagonist activity.
Coronavirus was previously shown to induce the unfolded-protein response (UPR) and ER stress in infected cell culture (32). Normally, ER is an active organelle for protein folding and modification. Loss of protein folding homeostasis would cause ER stress and induce the UPR, leading to the activation of three ER stress transducers. These transducers work in concert to attenuate translation and improve ER folding capacity to restore ER homeostasis (33). In this process, NF-κB is activated, and apoptosis will be induced if ER stress is prolonged (32, 33). In this study, we observed that the overexpression of ORFX protein led to cell death and the decrease of Renilla values (data not shown). This may imply that ORFX has a cytotoxic effect and an influence on overall protein translation. We also found that ORFX colocalizes with an ER marker. We hypothesize that ORFX may induce the UPR and cause ER stress which would activate NF-κB and induce apoptosis, promoting viral release at the late stage of infection.
It should be noted that the IFN and NF-κB detection systems used in this study were derived from and used in human cells. Since the innate immune system of bats is special and probably deficient in some aspects compared to the human system (34), it will be interesting to conduct the same studies in bat cells to determine whether ORFX protein has the same profiles as those observed in the human cell system. The development of different cell lines from the Rhinolophus bat, which is the reservoir host of SL-CoV, will facilitate this research in the future.
ACKNOWLEDGMENTS
We thank Hanzhong Wang, Zhenhua Zheng, Xianliang Ke, and Jin Meng (Research Group of Zoonotic Diseases, Wuhan Institute of Virology, CAS, China) for help and discussion, Yanyi Wang (Research Group of Molecular Immunology, Wuhan Institute of Virology, CAS, China) for kindly providing plasmids expressing cellular organelle markers (SecG1β-GFP, B4Gal-Ti-RFP, and Mito-YFP), Lin-Fa Wang (Duke-NUS Graduate Medical School, Singapore) for kindly providing a goat anti-SeV IgG, and Cecilia Waruhiu for language help.
This work was jointly funded by the National Natural Science Foundation of China (81290341, 31321001, and 81401672) and the National Institutes of Health (NIAID R01AI110964).
REFERENCES
1. Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT, Cheng VCC, Chan KH, Tsang DNC, Yung RWH, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi:10.1016/S0140-6736(03)13077-2. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
2. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538. doi:10.1038/nature12711. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
3. Yuan J, Hon CC, Li Y, Wang D, Xu G, Zhang H, Zhou P, Poon LL, Lam TT, Leung FC, Shi Z. 2010. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J Gen Virol 91:1058–1062. doi:10.1099/vir.0.016378-0. [PubMed] [CrossRef] [Google Scholar]
4. Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, Seebens A, Niedrig M, Pfefferle S, Yordanov S, Zhelyazkov L, Hermanns U, Vallo P, Lukashev A, Muller MA, Deng H, Herrler G, Drosten C. 2010. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol 84:11336–11349. doi:10.1128/JVI.00650-10. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
5. Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE. 2009. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis 15:482–485. doi:10.3201/eid1503.081013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. Lau SKP, Woo PCY, Li KSM, Huang Y, Tsoi HW, Wong BHL, Wong SSY, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A 102:14040–14045. doi:10.1073/pnas.0506735102. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Li WH, Zhang CS, Sui JH, Kuhn JH, Moore MJ, Luo SW, Wong SK, Huang IC, Xu KM, Vasilieva N, Murakami A, He YQ, Marasco WA, Guan Y, Choe HY, Farzan M. 2005. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24:1634–1643. doi:10.1038/sj.emboj.7600640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
8. He B, Zhang Y, Xu L, Yang W, Yang F, Feng Y, Xia L, Zhou J, Zhen W, Feng Y, Guo H, Zhang H, Tu C. 2014. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J Virol 88:7070–7082. doi:10.1128/JVI.00631-14. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Ren W, Li W, Yu M, Hao P, Zhang Y, Zhou P, Zhang S, Zhao G, Zhong Y, Wang S, Wang LF, Shi Z. 2006. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J Gen Virol 87:3355–3359. doi:10.1099/vir.0.82220-0. [PubMed] [CrossRef] [Google Scholar]
10. Yang XL, Hu B, Wang B, Wang MN, Zhang Q, Zhang W, Wu LJ, Ge XY, Zhang YZ, Daszak P, Wang LF, Shi ZL. 2015. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J Virol 90:3253–3256. [PMC free article] [PubMed] [Google Scholar]
11. Lau SK, Li KS, Huang Y, Shek CT, Tse H, Wang M, Choi GK, Xu H, Lam CS, Guo R, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol 84:2808–2819. doi:10.1128/JVI.02219-09. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Menachery VD, Yount BL Jr, Debbink K, Agnihothram S, Gralinski LE, Plante JA, Graham RL, Scobey T, Ge XY, Donaldson EF, Randell SH, Lanzavecchia A, Marasco WA, Shi ZL, Baric RS. 2015. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med 21:1508–1513. doi:10.1038/nm.3985. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. Menachery VD, Yount BL Jr, Sims AC, Debbink K, Agnihothram SS, Gralinski LE, Graham RL, Scobey T, Plante JA, Royal SR, Swanstrom J, Sheahan TP, Pickles RJ, Corti D, Randell SH, Lanzavecchia A, Marasco WA, Baric RS. 2016. SARS-like WIV1-CoV poised for human emergence. Proc Natl Acad Sci U S A doi:10.1073/pnas.1517719113. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
14. de Groot R, Baker S, Baric R, Enjuanes L, Gorbalenya A, Holmes K, Perlman S, Poon L, Rottier P, Talbot P, Woo P, Ziebuhr J. 2012. Family Coronaviridae, p 806–828. In King A, Adams M, Cartens E, Lefkowitz E (ed), Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, CA. [Google Scholar]
15. Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol 86:3995–4008. doi:10.1128/JVI.06540-11. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong SX, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. doi:10.1126/science.1085952. [PubMed] [CrossRef] [Google Scholar]
17. Li WD, Shi ZL, Yu M, Ren WZ, Smith C, Epstein JH, Wang HZ, Crameri G, Hu ZH, Zhang HJ, Zhang JH, McEachern J, Field H, Daszak P, Eaton BT, Zhang SY, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. doi:10.1126/science.1118391. [PubMed] [CrossRef] [Google Scholar]
18. Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, Denison MR, Davis N, Baric RS. 2005. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol 79:14909–14922. doi:10.1128/JVI.79.23.14909-14922.2005. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Liu DX, Fung TS, Chong KK-L, Shukla A, Hilgenfeld R. 2014. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res 109:97–109. doi:10.1016/j.antiviral.2014.06.013. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 81:548–557. doi:10.1128/JVI.01782-06. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
21. Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. 2009. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 4:e8342. doi:10.1371/journal.pone.0008342. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
22. Obitsu S, Ahmed N, Nishitsuji H, Hasegawa A, Nakahama K, Morita I, Nishigaki K, Hayashi T, Masuda T, Kannagi M. 2009. Potential enhancement of osteoclastogenesis by severe acute respiratory syndrome coronavirus 3a/X1 protein. Arch Virol 154:1457–1464. doi:10.1007/s00705-009-0472-z. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Kanzawa N, Nishigaki K, Hayashi T, Ishii Y, Furukawa S, Niiro A, Yasui F, Kohara M, Morita K, Matsushima K, Le MQ, Masuda T, Kannagi M. 2006. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett 580:6807–6812. doi:10.1016/j.febslet.2006.11.046. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
24. Zhou P, Li H, Wang H, Wang LF, Shi Z. 2012. Bat severe acute respiratory syndrome-like coronavirus ORF3b homologues display different interferon antagonist activities. J Gen Virol 93:275–281. doi:10.1099/vir.0.033589-0. [PubMed] [CrossRef] [Google Scholar]
25. Ren W, Qu X, Li W, Han Z, Yu M, Zhou P, Zhang SY, Wang LF, Deng H, Shi Z. 2008. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J Virol 82:1899–1907. doi:10.1128/JVI.01085-07. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
26. Perrotta AT, Been MD. 1991. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature 350:434–436. doi:10.1038/350434a0. [PubMed] [CrossRef] [Google Scholar]
27. Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J 22:2552–2560. doi:10.1093/emboj/cdg267. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
28. DeDiego ML, Nieto-Torres JL, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, Usera F, Enjuanes L. 2014. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res 194:124–137. doi:10.1016/j.virusres.2014.07.024. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Almazan F, Gonzalez JM, Penzes Z, Izeta A, Calvo E, Plana-Duran J, Enjuanes L. 2000. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A 97:5516–5521. doi:10.1073/pnas.97.10.5516. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
30. Yount B, Curtis KM, Baric RS. 2000. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J Virol 74:10600–10611. doi:10.1128/JVI.74.22.10600-10611.2000. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Donaldson EF, Sims AC, Baric RS. 2008. Systematic assembly and genetic manipulation of the mouse hepatitis virus A59 genome. Methods Mol Biol 454:293–315. doi:10.1007/978-1-59745-181-9_21. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
32. Fung TS, Liu DX. 2014. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol 5:296. doi:10.3389/fmicb.2014.00296. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
33. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d' Hellencourt C, Ravanan P. 2014. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci 8:213. doi:10.3389/fncel.2014.00213. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
34. Baker ML, Schountz T, Wang LF. 2013. Antiviral immune responses of bats: a review. Zoonoses Public Health 60:104–116. doi:10.1111/j.1863-2378.2012.01528.x. [PMC free article] [PubMed] [CrossRef] [Google Scholar]