Lab-Made? SARS-CoV-2 Genealogy Through the Lens of Gain-of-Function Research
by Yuri Deigin
Apr 22, 2020
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.

Staff celebrating the physical completion of the laboratory in 2015, Wuhan, China (Source)
If you hear anyone claim “we know the virus didn’t come from a lab”, don’t buy it — it may well have. Labs around the globe have been creating synthetic viruses like CoV2 for years. And no, its genome would not necessarily contain hallmarks of human manipulation: modern genetic engineering tools permit cutting and pasting genomic fragments without leaving a trace. It can be done quickly, too: it took a Swiss team less than a month to create a synthetic clone of CoV2.
How I Learned to Start Worrying
Oh, come on. Lab-made? Nonsense! Back in January, that was my knee-jerk reaction when ideas that Covid-19 is caused by a laboratory leak had just surfaced. Bioweapon? Well, that is just Flat Earth crazies territory. Thus, whenever I kept hearing anything about non-natural origins of SARS-CoV-2, I brushed it aside under similar sentiments. So what if there is a virology institute in Wuhan? Who knows how many of those are sprinkled throughout China.
At some point, it became necessary to brush such theories aside in a substantiated manner, as their proponents began to back up their theses about the possible artificial nature of the virus with arguments from molecular biology, and when engaging them in debate, I wanted to smash their conspiracy theories with cold, hard scientific facts. Just like that Nature paper (or so I thought).
So it was then, in pursuit of arguments against the virus’s lab-madeness, that I got infected by the virus of doubt. What was the source of my doubts? The fact that the deeper you dive into the research activities of coronavirologists over the past 15–20 years, the more you realize that creating chimeras like CoV2 was commonplace in their labs.
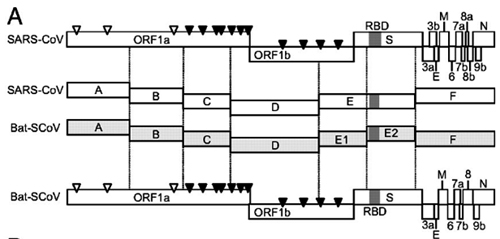
A chimera virus is defined by the Center for Veterinary Biologics (part of the U.S. Department of Agriculture's Animal and Plant Health Inspection Service) as a "new hybrid microorganism created by joining nucleic acid fragments from two or more different microorganisms in which each of at least two of the fragments contain essential genes necessary for replication." The term chimera already referred to an individual organism whose body contained cell populations from different zygotes or an organism that developed from portions of different embryos. In mythology, a chimera is a creature such as a hippogriff or a gryphon formed from parts of different animals, thus the name for these viruses. Chimeric flaviviruses have been created in an attempt to make novel live attenuated vaccines.
-- Chimera (virus), by Wikipedia
And CoV2 is an obvious chimera (though not necessarily a lab-made one), which is based on the ancestral bat strain RaTG13, in which the receptor binding motif (RBM) in its spike protein is replaced by the RBM from a pangolin strain, and in addition, a small but very special stretch of 4 amino acids is inserted, which creates a furin cleavage site that, as virologists have previously established, significantly expands the “repertoire” of the virus in terms of whose cells it can penetrate. Most likely, it was thanks to this new furin site that the new mutant managed to jump species from its original host to humans.
Indeed, virologists, including the leader of coronavirus research at the Wuhan Institute of Virology, Shi Zhengli, have done many similar things in the past — both replacing the RBM in one type of virus by an RBM from another, or adding a new furin site that can provide a species-specific coronavirus with an ability to start using the same receptor (e.g. ACE2) in other species. In fact, Shi Zhengli’s group was creating chimeric constructs as far back as 2007 and as recently as 2017, when they created a whole of 8 new chimeric coronaviruses with various RBMs. In 2019 such work was in full swing, as WIV was part of a $3.7 million NIH grant titled Understanding the Risk of Bat Coronavirus Emergence. Under its auspices, Shi Zhengli co-authored a 2019 paper that called for continued research into synthetic viruses and testing them in vitro and in vivo:
Currently, no clinical treatments or prevention strategies are available for any human coronavirus. Given the conserved RBDs of SARS-CoV and bat SARSr-CoVs, some anti-SARS-CoV strategies in development, such as anti-RBD antibodies or RBD-based vaccines, should be tested against bat SARSr-CoVs. Recent studies demonstrated that anti-SARS-CoV strategies worked against only WIV1 and not SHC014. In addition, little information is available on HKU3-related strains that have much wider geographical distribution and bear truncations in their RBD. Similarly, anti-S antibodies against MERS-CoV could not protect from infection with a pseudovirus bearing the bat MERSr-CoV S. Furthermore, little is known about the replication and pathogenesis of these bat viruses. Thus, future work should be focused on the biological properties of these viruses using virus isolation, reverse genetics and in vitro and in vivo infection assays. The resulting data would help the prevention and control of emerging SARS-like or MERS-like diseases in the future.
If the above quote might seem vague as to what exactly “using reverse genetics” might mean, the NIH grant itself spells it out:
Aim 3. In vitro and in vivo characterization of SARSr-CoV spillover risk, coupled with spatial and phylogenetic analyses to identify the regions and viruses of public health concern. We will use S protein sequence data, infectious clone technology, in vitro and in vivo infection experiments and analysis of receptor binding to test the hypothesis that % divergence thresholds in S protein sequences predict spillover potential.
“Infectious clone technology” stands for creating live synthetic viral clones. Considering the heights of user friendliness and automation that genetic engineering tools have attained, creating a synthetic CoV2 via the above methodology would be in reach of even a grad student.
But before delving into CoV2 origins, let’s first take a quick dive into its biology.
Biology
Ok, let’s start from the basics. What’s a furin site, an RBM, or a spike protein? Bear with me: once you wade through the jungle of terminology, conceptually, everything is pretty straightforward. For example, spike proteins are those red things sticking out of a virus particle — the very reason for which these viruses got “crowned”:
It is with the help of these proteins that the virion clings to the receptor of the victim cell (ACE2 in our case) to then penetrate inside. So it is a vitally important part of the virus, as without getting into a cell viruses cannot replicate. The spike protein also determines which animals the virus can or cannot infect, as ACE2 receptors (or other targets for other viruses) in different species can differ in structure. At the same time, out of the entire 30 kilobase genome (quite huge by viral standards), the gene of this protein makes up only 12–13%. So the spike protein is only about 1300 amino acids long. Below is how the spike (S) protein is structured in CoV2 and close relatives:
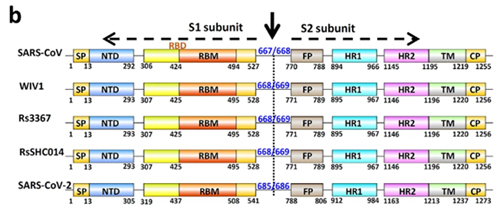
As can be seen from the figure above, the S protein consists of two subunits: S1 and S2. It is S1 that interacts with the ACE2 receptor, and the place where S1 does so is called Receptor Binding Domain (RBD), while the area of direct contact, the holy of holies, is called Receptor Binding Motif (RBM). Here is a beautiful illustration from an equally beautiful work:
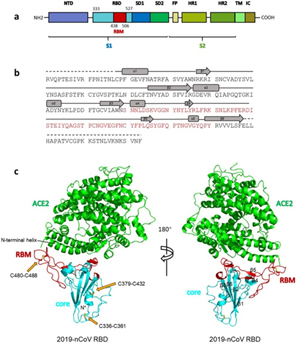
Overall structure of 2019-nCoV RBD bound with ACE2.
(a) Overall topology of 2019-nCoV spike monomer. NTD, N-terminal domain. RBD, receptor-binding domain. RBM, receptor-binding motif. SD1, subdomain 1. SD2, subdomain 2. FP, fusion peptide. HR1, heptad repeat 1. HR2, heptad repeat 2. TM, transmembrane region. IC, intracellular domain.
(b) Sequence and secondary structures of 2019-nCoV RBD. The RBM is colored red.
© Overall structure of 2019-nCoV RBD bound with ACE2. ACE2 is colored green. 2019-nCoV RBD core is colored cyan and RBM is colored red. Disulfide bonds in the 2019-nCoV RBD are shown as stick and indicated by yellow arrows. The N-terminal helix of ACE2 responsible for binding is labeled.
When the CoV2 genome was just sequenced and made publicly available on January 10, 2020, it was a riddle, as no closely related strains were known. But quite quickly, on January 23, Shi Zhengli released a paper indicating that CoV2 is 96% identical to RaTG13, a strain which her laboratory had previously isolated from Yunnan bats in 2013. However, outside of her lab, no one knew about that strain until January 2020.
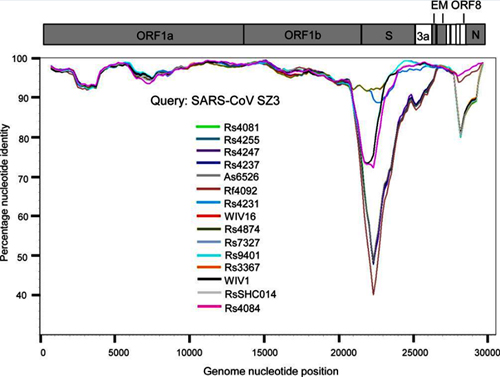
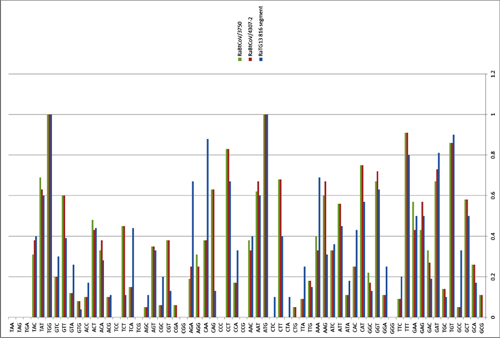
It was immediately clear that RaTG13 is special. Take a look at the figure below:
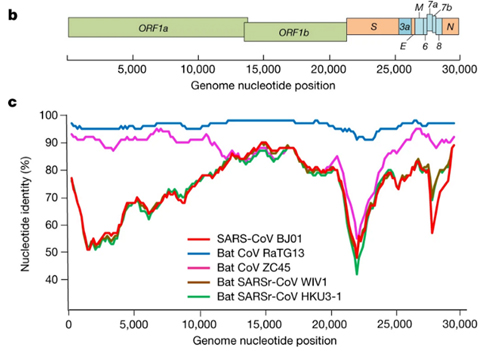
This is a genome similarity graph between CoV2 and other known strains. The higher the curve, the higher the percentage of matching nucleotides. As you can see, in the spike protein (S) gene region (between nucleotides 22k and 25k), only RaTG13 is more or less close to CoV2, while all other strains take a deep dive around this spot — both strains from other bats and the first SARS-CoV (red curve). This in itself is far from suspicious — who knows how many unknown SARS-like strains lurk in the bat caves of Yunnan? Ok, maybe it is not very clear how exactly the virus could get from there to Wuhan, but hey, with those wet markets you never know.
Pangolins
Next, pangolins appeared on the scene: in February, another group of Chinese scientists discovered a peculiar strain of pangolin coronavirus in their possession, which, while generally being only 90% similar to CoV2, in the RBM region was almost identical to it, with only a single amino acid difference (see the upper two sequences, dots indicate a match with the top sequence):

Surprisingly, in the first quarter of the S protein, the pangolin strain is highly dissimilar from CoV2, but after the RBM all three strains (CoV2, Pangolin, RaTG13) exhibit a shared high degree of similarity. Most strikingly, RaTG13’s RBM itself is quite different than that of CoV2, which can be seen from the steep dive of the green RaTG13 graph compared to the red CoV2 graph in the RBM region (pink strip) in the following graph:
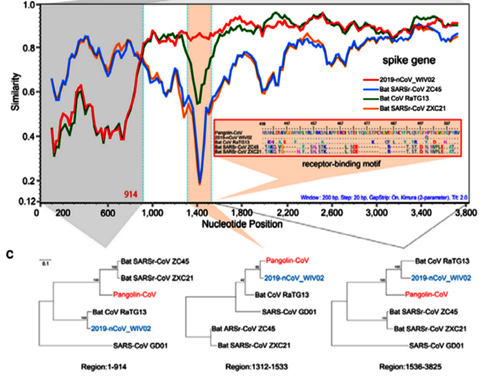
This observation is confirmed by the phylogenetic analysis of the three areas highlighted in the graph above — in the RBM, the pangolin strain is closer to CoV2 than is RaTG13, but it is RaTG13 that is closer to CoV2 to the left and right of RBM. So there is obvious recombination, as the authors (and other papers) conclude.
Genetic recombination (also known as genetic reshuffling) is the exchange of genetic material between different organisms which leads to production of offspring with combinations of traits that differ from those found in either parent.
-- Genetic recombination, by Wikipedia
How did the researchers obtain those pangolins? This is how:

They were confiscated from smugglers by Chinese customs and transferred to an animal rehab center in Guangdong, where they died while exhibiting severe coronavirus symptoms. This, of course, must have gotten the attention of local virologists, who took several samples:
Pangolins used in the study were confiscated by Customs and Department of Forestry of Guangdong Province in March-December 2019. They include four Chinese pangolins (Manis pentadactyla) and 25 Malayan pangolins (Manis javanica). These animals were sent to the wildlife rescue center, and were mostly inactive and sobbing, and eventually died in custody despite exhausting rescue efforts. Tissue samples were taken from the lung, lymph nodes, liver, spleen, muscle, kidney, and other tissues from pangolins that had just died for histopathological and virological examinations.
Those pangolins attracted the attention of other virologists too. For example, a team in Hong Kong also received samples of confiscated pangolins and in February 2020 they also released a paper that noted clear signs of recombination in the CoV2 spike protein:
We received frozen tissue (lungs, intestine, blood) samples that were collected from 18 Malayan pangolins (Manis javanica) during August 2017-January 2018. These pangolins were obtained during the anti-smuggling operations by Guangxi Customs. Strikingly, high-throughput sequencing of their RNA revealed the presence of coronaviruses in six (two lung, two intestine, one lung-intestine mix, one blood) of 43 samples. With the sequence read data, and by filling gaps with amplicon sequencing, we were able to obtain six full or nearly full genome sequences — denoted GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L — that fall into the 2019-CoV2 lineage (within the genus Betacoronavirus) in a phylogenetic analysis (Figure 1a).
…
More notable, however, was the observation of putative recombination signals between the pangolins coronaviruses, bat coronaviruses RaTG13, and human 2019-CoV2 (Figure 1c, d). In particular, 2019-CoV2 exhibits very high sequence similarity to the Guangdong pangolin coronaviruses in the receptor-binding domain (RBD; 97.4% amino acid similarity; indicated by red arrow in Figure 1c and Figure 2a), even though it is most closely related to bat coronavirus RaTG13 in the remainder of the viral genome. Bat CoV RaTG and the human 2019-CoV2 have only 89.2% amino acid similarity in RBD. Indeed, the Guangdong pangolin coronaviruses and 2019-CoV2 possess identical amino acids at the five critical residues of the RBD, whereas RaTG13 only shares one amino acid with 2019-CoV2 (residue 442, human SARS-CoV numbering).
By the way, the authors of this article also highlighted the high phylogenetic mosaicity of the CoV2 spike protein:
Interestingly, a phylogenetic analysis of synonymous sites alone in the RBD revealed that the phylogenetic position of the Guangdong pangolin is consistent with that in the remainder of the viral genome, rather than being the closest relative of 2019-CoV2 (Figure 2b). Hence, it is possible that the amino acid similarity between the RBD of the Guangdong pangolin coronaviruses and 2019-CoV2 is due to selectively-mediated convergent evolution rather than recombination, although it is difficult to choose between these scenarios on current data.
Translated from science-speak, what this means is that if we analyze the entire RBD of the three strains, ignoring the obvious differences (i.e. non-synonymous substitutions) among them, which are mainly found in the RBM (which, recall, is identical between CoV2 and Pangolin), and construct a phylogenetic tree for synonymous substitutions, CoV2 is still closer to RaTG13 than to the pangolin strain. Which is rather strange in light of the fact that the pangolin strain and CoV2 have identical RBMs (which are segments inside RBD).
The authors go on to put forth a conjecture that this may be the result of convergent evolution, in other words, that CoV2 and the pangolin strain came to possess identical RBMs each in their own way, rather than through recombination between common ancestors. Because it would have required a rather unique recombination event — as if someone cut out a precise RBM segment from a pangolin strain and used it to replace the RBM in RaTG13. Talk about Intelligent Design!
Royal Genealogy
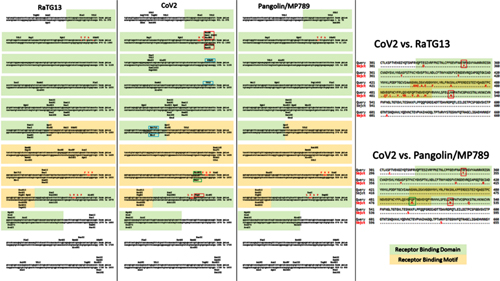
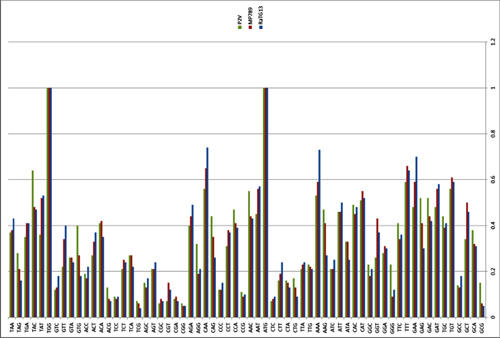
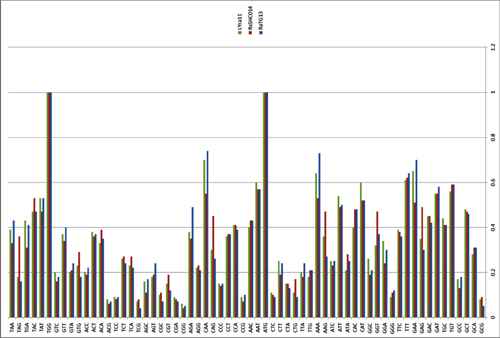
In order to better understand CoV2 origins, let’s take a look at spike protein sequences of our Unholy Trinity: CoV2, RaTG13 and MP789 (pangolin-2019). Let’s compare the pairwise differences between them (identical amino acids are marked with dots, red letters denote differences, and dashes indicate deleted/inserted amino acids):

The comparisons illustrate what previously quoted papers have noted: that in the first quarter of the sequence, the pangolin strain is far from CoV2 and RaTG1, and if it weren’t for the RBM region (red rectangle), RaTG13 would have been very close to CoV2. But, as I already said, the RBM in CoV2 is closest to that of the pangolin strain.
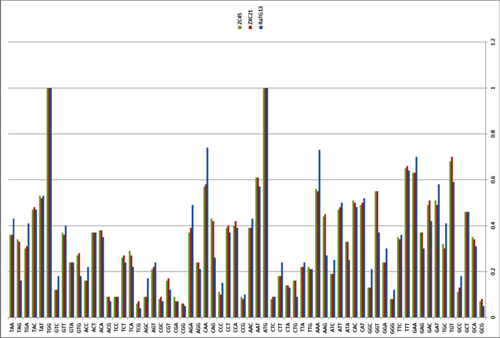
What about other pangolin strains? So far we’ve only analyzed the MP789 strain isolated from pangolins confiscated by customs in 2019. But there was another batch of pangolins confiscated in 2017, and they also had a similar coronavirus strain isolated. Let’s compare it to RaTG13 and MP789:

In the first quarter of the S protein, the 2017 pangolin strains are closer to RaTG13 (and CoV2) than their 2019 pangolin counterpart (MP789). At the same time, all three have a clear recent common ancestor in the areas marked by green rectangles, and in these areas RaTG13 and pangolin-2019 (MP789) are closer to each other than to pangolin-2017, since they have several common mutations (marked by red and blue ellipses), which are absent from pangolin-2017. But the RBM for all three is different, and different in approximately the same proportion, and in similar places.
Maybe after ancestors of RaTG13 and MP789 diverged, the MP789 ancestor had the first quarter of its protein replaced (which did not occur in RaTG13 or pangolin-2017), and the rest of the protein remained common for all three strains. Later the paths of the RaTG13 and MP789 gene pools crossed again and produced CoV2. It is also possible that the ancestor of RaTG13 arose as a result of recombination of ancestral pangolin strains.
It is also interesting to see a rather unique identical mutation (QTQTNS) in RaTG13 and pangolin-2019 right in front of the spot where CoV2 has a new furin cleavage site. That furin site, as I mentioned, arose via an insertion of 4 new amino acids (PRRA). If we look at the nucleotide sequence around this insertion, we can see that RaTG13 and CoV2 are closer to each other in that area than to pangolin-2019, since they possess several common mutations (highlighted in blue):

By the way, Orf1ab is also a phylogenetic mess in CoV2: 1a is closer to RaTG13, but 1b is closer to pangolin-2019:
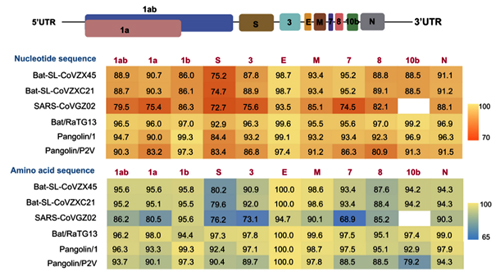
(Image Source)
Does this mean that the ancestor of CoV2 crossed with the common ancestor of pangolin-19 at least twice? First, when it (along with a common ancestor of RaTG13) inherited Orf1ab and the second half of the spike protein with the QTQTNS mutation, and second time when it acquired 1b and RBM, which differ from RaTG13. All of this is certainly possible in nature — after all, these viruses mutate and recombine constantly. Another question is where exactly bat and pangolin viruses are most likely to encounter one another for such orgies — in mountain caves, “wet markets”, shelters for confiscated animals, or even in laboratories. But let’s put those questions aside for now. First, let's discuss what is arguably the most eye-catching aspect of the new virus — a 4-amino acid insertion that turned it into a natural-born killer.
A Killer Intro
It is impossible to ignore the introduction of a PRRA insert between S1 and S2: it sticks out like a splinter. This insert creates the furin cleavage site, which I mentioned at the very beginning. Let me explain what a furin site is. Remember the structure of our spike protein? Here is a detailed diagram:
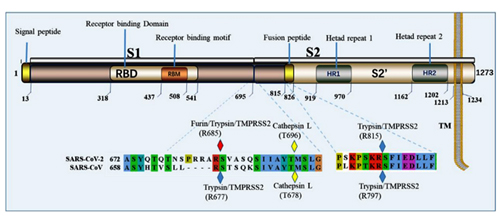
The protein consists of two parts, S1 and S2, of which S1 is responsible for primary contact with the receptor (recall Receptor Binding Domain / Motif), and S2 is responsible for fusion with the cell membrane and penetration into the cell. The fusion process is started by the fusion peptide marked in yellow, but in order for it to engage in its dirty deed, someone must cut the S protein at one of the sites marked by diamonds in the diagram above. The virus does not have its own such “cutters”, so it relies on various proteases of its victims. There are several types of such proteases, as can be deduced from the abundance of colors of those diamonds. But not all proteases are equal, and not all types of cells have proteases needed by the virus. Furin is one of the most effective, and it is found not only on the surface of cells, but also inside. Most clearly, the danger of the new furin site is demonstrated by the difference between CoV2 and its grandpa, SARS-CoV:
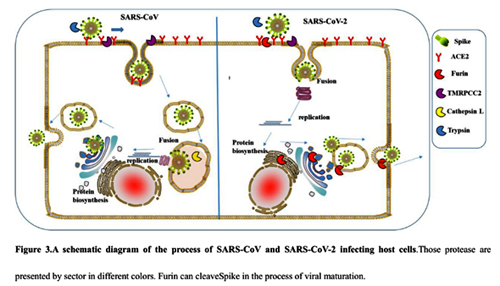
As can be seen from the diagram, in the case of CoV2, thanks to the furin site, it is not two, but three classes of proteases (three colored PacMans) that can cut its S protein outside the cell. But perhaps the most important difference is that furin is also present inside the cell, so it can cut the S protein immediately after virion assembly, thereby providing new virions with the ability to merge with new cells right off the bat (no pun intended).
The importance of the new furin site in CoV2’s virulence was recently demonstrated by a study in hamsters where the disappearance of the furin site (due to a mutation) greatly decreased mutant CoV2’s pathogenicity and replication ability:
Infection of hamsters shows that one of the variants (Del-mut-1) which carries deletion of 10 amino acids (30 bp) does not cause the body weight loss or more severe pathological changes in the lungs that is associated with wild type virus infection.
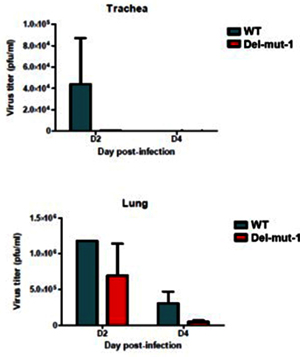
Virus replication in the lung tissues of hamsters infected with either WT or Del-mut-1 SARS-CoV-2 virus. Virus titration by plaque assay of lung and tracheal tissues collected on day 2 and 4 post-infection
The good news is that there already exist various furin and other protease inhibitors, and some of them (like camostat and its analogs) are already being clinically tested against CoV2.
By the way, it is possible that the new furin site could also be largely responsible for the pronounced age-dependent morbidity and mortality of CoV2:
Patients with hypertension, diabetes, coronary heart disease, cerebrovascular illness, chronic obstructive pulmonary disease, and kidney dysfunction have worse clinical outcomes when infected with SARS-CoV-2, for unknown reasons. The purpose of this review is to summarize the evidence for the existence of elevated plasmin(ogen) in COVID-19 patients with these comorbid conditions. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS-CoV-2, extracellularly, which increases its infectivity and virulence.
Plasmin is an important enzyme (EC 3.4.21.7) present in blood that degrades many blood plasma proteins, including fibrin clots...
Plasmin is a serine protease that acts to dissolve fibrin blood clots. Apart from fibrinolysis, plasmin proteolyses proteins in various other systems: It activates collagenases, some mediators of the complement system, and weakens the wall of the Graafian follicle, leading to ovulation. It cleaves fibrin, fibronectin, thrombospondin, laminin, and von Willebrand factor. Plasmin, like trypsin, belongs to the family of serine proteases.
-- Plasmin, by Wikipedia
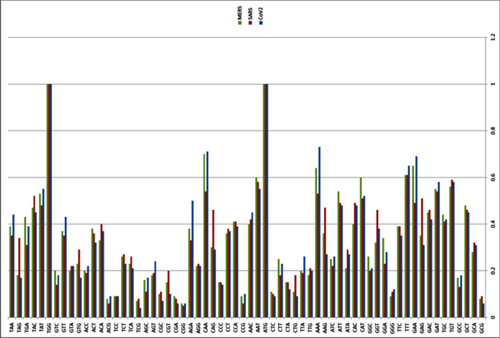
Furin cuts proteins in strictly defined places, namely after an RxxR sequence (that is, Arg-X-X-Arg, where X can be any amino acid). Moreover, if arginine is also in the second or third place (that is, RRxR or RxRR), then the cleavage efficiency is significantly increased.
Therefore, the appearance of a new furin cleavage site was noticed immediately, as none of the closest or even distant relatives of Cov2 have such a site — those coronaviruses that do, share only 40% of their genome with Cov2:
It was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site “RRAR” in SARS-CoV-2 is unique in its family, rendering by its unique insert of “PRRA”. The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
Here is a great illustration from the source article of the quote above. Coronaviruses with a furin site are marked in pink, 3 different strains of Cov2 are shown at 10 o’clock:
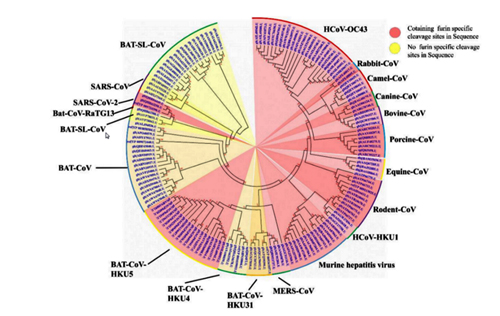
The closest relative with a furin site is the HKU5 strain, isolated by the Shi Zhengli team in 2014 in Guangzhou from bats of the genus Pipistrellus (added to GenBank in 2018). But it is a very distant relative — their spike proteins share only 36%.
So the virologists are puzzled. Where did this 12 nucleotide insert come from? Could it be lab-made? Well, virologists have studied furin sites in coronaviruses for decades, and have introduced many artificial ones in a lab. For example, an American team had inserted RRSRR into the spike protein of the first SARS-CoV back in 2006:
To investigate whether proteolytic cleavage at the basic amino acid residues, were it to occur, might facilitate cell–cell fusion activity, we mutated the wild-type SARS-CoV glycoprotein to construct a prototypic furin recognition site (RRSRR) at either position.

And the Japanese have inserted a similar site (RRKR) into the SARS-CoV protein in 2008, though a bit downstream than in CoV2:
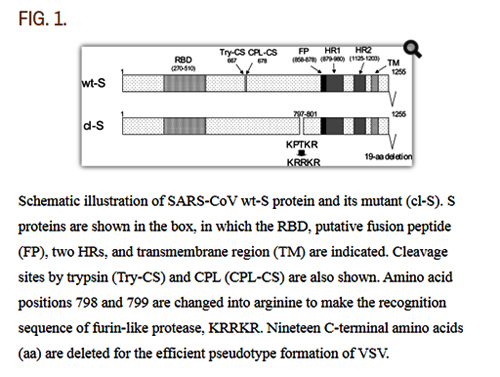
Schematic illustration of SARS-CoV wt-S protein and its mutant (cl-S). S proteins are shown in the box, in which the RBD, putative fusion peptide (FP), two HRs, and transmembrane region (TM) are indicated. Cleavage sites by trypsin (Try-CS) and CPL (CPL-CS) are also shown. Amino acid positions 798 and 799 are changed into arginine to make the recognition sequence of furin-like protease, KRRKR. Nineteen C-terminal amino acids (aa) are deleted for the efficient psuedotype formation of VSV.
In the same year 2008, their Dutch colleagues also studied these protease sites of SARS-CoV and compared them to the murine coronavirus MHV, which also has such a site (SRRAHR | SV), one that is quite similar to the site of CoV2 (SPRRAR | SV):
In 2009, another American group also worked on “improving” SARS-CoV and, continuing the American tradition of not penny-pinching on arginines, they inserted as many as 4 of them (RRSRR):
To examine the potential use of the SARS-CoV S1–S2 and S2′ positions as sites for proteolytic cleavage, we first introduced furin cleavage recognition sites at these locations by making the following mutations 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1–S2) and 792-LKPTKRSF — LKRTKRSF-799 (S2′).
Beijing 2019
But the most recent work of this kind that I came across was an October 2019 paper from several Beijing labs, where the new furin site RRKR was inserted into not just some pseudovirus, but into an actual live chicken coronavirus, infectious bronchitis virus (IBV):
An interesting side note is that, as the authors point out, the addition of a furin site allows the mutant virus to infect nerve cells. Perhaps the CoV2 furin site is the reason why some patients with CoV2 exhibit neurological symptoms, including loss of smell:
Mutation of the S2' site of QX genotype (QX-type) spike protein (S) in a recombinant virus background results in higher pathogenicity, pronounced neural symptoms and neurotropism when compared with conditions in wild-type IBV (WT-IBV) infected chickens. In this study, we present evidence suggesting that recombinant IBV with a mutant S2' site (furin-S2' site) leads to higher mortality. Infection with mutant IBV induces severe encephalitis and breaks the blood–brain barrier.
…
In summary, our results demonstrate that the furin cleavage site upstream of the FP in S protein is an important site for CoV, modulating entry, cell–virus fusion, adaptation to its host cell, cell tropism and pathogenicity, but not antigenicity.
Encephalitis is inflammation of the brain. There are several causes, but the most common is a viral infection.
Encephalitis often causes only mild flu-like signs and symptoms — such as a fever or headache — or no symptoms at all. Sometimes the flu-like symptoms are more severe. Encephalitis can also cause confused thinking, seizures, or problems with movement or with senses such as sight or hearing.
In some cases, encephalitis can be life-threatening. Timely diagnosis and treatment are important because it's difficult to predict how encephalitis will affect each individual.
Encephalitis, by Mayo Clinic
To be clear, many coronaviruses have naturally occurring furin sites, and they are very diverse. Obviously, they can appear as a result of random mutations. This is what happened in the case of MERS, as was pointed out in 2015 by an international team of authors, including Shi Zhengli and Ralph Baric, two stars of synthetic coronavirusology. We will come back to them many times, but for now, a few words about that article. In it the authors have shown that just two mutations allowed MERS to jump from bats to humans, and one of these mutations created a furin site. Though it was not an insertion of new amino acids, but a mutation of an existing one (marked in red on the left below):
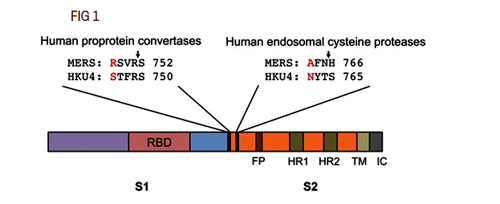
The authors did not just show this, but actually introduced these mutations back into the original bat strain: they created the same furin site and showed that it enables the bat strain to infect human cells:
To evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
…
We examined the capability of the mutant HKU4 spike to mediate viral entry into three types of human cells (Fig. 3A for HEK293T cells; data not shown for Huh-7 and MRC-5 cells), using a pseudovirus entry assay as previously described (14). In the absence of exogenous protease trypsin, HKU4 pseudoviruses bearing either the reengineered hPPC motif or the reengineered hECP motif were able to enter human cells, whereas HKU4 pseudoviruses bearing both of the reengineered human protease motifs entered human cells as efficiently as when activated by exogenous trypsin (Fig. 3A). In contrast, wild-type HKU4 pseudoviruses failed to enter human cells. Therefore, the reengineered hPPC and hECP motifs enabled HKU4 spike to be activated by human endogenous proteases and thereby allowed HKU4 pseudoviruses to bypass the need for exogenous proteases to enter human cells. These results reveal that HKU4 spike needs only two single mutations at the S1/S2 boundary to gain the full capacity to mediate viral entry into human cells.
By the way, how they did it might frighten those who aren’t familiar with modern biotechnology — because the authors inserted this coronavirus spike-like protein into inactivated HIV:
Briefly, MERS-CoV-spike-pseudotyped retroviruses expressing a luciferase reporter gene were prepared by cotransfecting HEK293T cells with a plasmid carrying Env-defective, luciferase-expressing HIV-1 genome (pNL4–3.luc.R-E-) and a plasmid encoding MERS-CoV spike protein.
Perhaps this is what prompted Indian researchers to look for sequences similar to HIV in the CoV2 genome (but their preprint was quickly criticized for bad methodology and erroneous conclusions). In fact, experts use such pseudoviruses regularly, and in general, one should not be scared of retroviruses as a class — their subspecies lentiviruses have been used for gene therapy for many years.
Where Did RaTG13 Come From?
RaTG13 is a very unusual strain. Odd to see that Shi Zhengli’s group was silent about it for all these years. After all, it is very different from its SARS-like siblings, especially in the spike protein, which is precisely what determines which types of cells (and in which animals) this virus can infect. Here is a genome similarity graph of CoV2 compared to other bat coronaviruses (panel B):
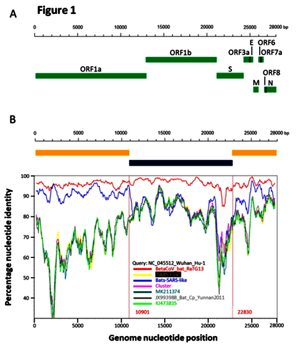
The red curve represents RaTG13 while the blue curve is for the strains closest to RaTG13 (ZXC21 and ZC45). These strains were isolated from Chinese horseshoe bats (Rhinolophus sinicus) in Zhoushan in 2015 (ZXC21) and 2017 (ZC45). As can be seen from the above graph, even they differ in their S proteins from RaTG13. A direct sequence comparison illustrates this difference best:

As we can see, the spike proteins of ZXC21 and ZC45 are not only 23–24 amino acid residues shorter than the RaTG13 protein, but they are shorter in the most important place — in the RBM (note the deletions in the red box marked with red dashes).
So where did RaTG13 come from? As I already mentioned, in 2020 Shi Zhengli reported that she isolated it in 2013 from Yunnan horseshoe bats (from Rhinolophus affinis, not the usual suspects R. sinicus). But until January 2020, this strain’s existence was not known, and here is how Shi Zhengli’s group described their discovery about RaTG13’s similarity to CoV2:
We then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13) — which was previously detected in Rhinolophus affinis from Yunnan province — showed high sequence identity to 2019-CoV2. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-CoV2 was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%.
Not much detail: previously detected, and that is that. Moreover, the quote seems to imply that until 2020, they only sequenced a part of its genome, the RdRp gene (which is part of Orf1b that precedes the spike protein gene). Ok, but where exactly in Yunnan was it obtained? The paper doesn’t mention it, and neither does GenBank. However, the GISAID entry seems to have a bit more info: collected in Pu’er City from a male bat’s fecal swab:

This rang a bell, as in my wanderings around Pubmed, I had already encountered an expedition to Pu’er in the summer of 2013:
Bats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013.
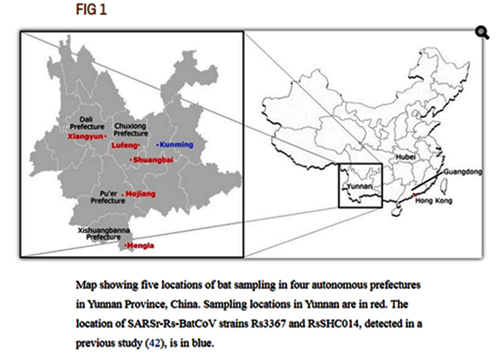
Map showing five locations of bat sampling in four autonomous prefectures in Yunnan Province, China. Sampling locations in Yunnan are in red. The location of SARSr-Rs-BatCoV strains Rs3367 and RsSHC014, detected in a previous study (42), is in blue.
Researchers did not report anything particularly interesting for us from that expedition, but maybe it was then that Shi Zhengli or someone from her group obtained the RaTG13 sample? Which they sequenced only partially, and for some reason decided not to publish, although it was very different from everything known before.
By the way, Shi Zhengli could well have personally participated in that expedition, as she expressed great fondness when describing them — for example, in her TED-like talk in 2018, where she showed personal photos from such expeditions:
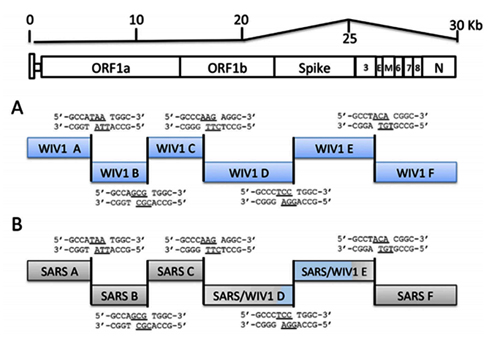
CREATOR OF NEW CORONAVIRUS? WUHAN INSTITUTE OF VIROLOGY
Moreover, it was a series of exactly such expeditions that brought Shi Zhengli worldwide fame and a “Batwoman” moniker: in a 2013 Nature paper, her group triumphantly announced that in Yunnan caves they had discovered carrier bats of the RsSHC014 and Rs3367 strains that coincided with the first SARS-CoV by 85% and 96%, respectively.
It is quite a coincidence that around the same time in Yunnan, Shi Zhengli’s group also discovered RaTG13, the closest strain to CoV2, and the two also share 96% of their genomes.
UPD: Is RaTG13 the same as RaBtCoV/4991?
[UPDATED] After I had published this post, I was pointed to this preprint that alleges that RaTG13 is, in fact, RaBtCoV/4991 (KP876546), which Shi Zhengli had previously reported discovering in an abandoned mineshaft in Yunnan in 2013. There indeed are several reasons to think so. First and foremost, the only published sequence for RaBtCoV/4991 is 100% identical to that of RaTG13 at the nucleotide level, albeit being just a 370-bp stretch of the RdRp gene:
BtCoV/4991 was first described in 2016. It is a 370 nucleotide virus fragment collected from the Mojiang mine in 2013 by the lab of Zeng-li Shi at the WIV [Wuhan Institute of Virology] (Ge et al., 2016). BtCoV/4991 is 100% identical in sequence to one segment of RaTG13. RaTG13 is a complete viral genome sequence (almost 30,000 nucleotides) that was only published in 2020, after the pandemic began (P. Zhou et al., 2020).
Despite the confusion created by their different names, in a letter obtained by us Zheng-li Shi confirmed to a virology database that BtCoV/4991 and RaTG13 are both from the same bat faecal sample and the same mine. They are thus sequences from the same virus...
Why did the Shi lab not acknowledge the miners’ deaths in any paper describing samples taken from the mine (Ge et al., 2016 and P. Zhou et al., 2020)? Why in the title of the Ge at al. 2016 paper did the Shi lab call it an “abandoned” mine? When they published the sequence of RaTG13 in Feb. 2020, why did the Shi lab provide a new name (RaTG13) for BtCoV/4991 when they had by then cited BtCoV/4991 twice in publications and once in a genome sequence database and when their sequences were from the same sample and 100% identical (P. Zhou et al., 2020)? If it was just a name change, why no acknowledgement of this in their 2020 paper describing RaTG13 (Bengston, 2020)? These strange and unscientific actions have obscured the origins of the closest viral relatives of SARS-CoV-2, viruses that are suspected to have caused a COVID-like illness in 2012 and which may be key to understanding not just the origin of the COVID-19 pandemic but the future behaviour of SARS-CoV-2.
-- A Proposed Origin for SARS-CoV-2 and the COVID-19 Pandemic [W/Comments], by Jonathan Latham, PhD and Allison Wilson, PhD

Second, the collection details of the two strains are nearly identical: both were collected in July 2013 from a fecal swab of R. affinis bats:

RaBtCoV/4991 was collected in a mineshaft located in the Mojiang county, which is under the jurisdiction of Pu’er City:
Mojiang Hani Autonomous County is an autonomous county under the jurisdiction of Pu’er City, in the south of Yunnan Province, China.
-- Wikipedia
And Pu’er City is listed as the collection location of RaTG13 at the GISAID database, which could well be an approximation for the Mojiang mineshaft.
It is odd that in her 2020 paper on RaTG13 Shi Zhengli fails to mention RaBtCoV/4991 or cite her 2016 paper about its discovery, for which she is listed as the one who “designed and coordinated the study”. It is not like RaBtCoV/4991 was forgotten by her group, as it is mentioned in their 2019 paper, where it is included in a phylogenetic tree of other coronaviruses:

Sampling map (A) and phylogenetic analysis of CoVs detected in Rhinolophus bats (B). A total of 19 provinces (indicated in gray) in China were involved. 1. Beijing (BJ), 2. Chongquing (CA); 3. Fujian (FJ); 4. Gansu (GS); 5. Guangdong (GD); 6. Guangxi (BX); 7. Guizhou (GZ); 8. Hainan (HaN); 9. Hebei (HeB); 10. Henan (HeN); 11. Hubei (HuB); 12. Hunan (HuN); 13. Jiangsu (JS); 14. Shandong (SD); 15. Shanxi (SX); 16. Sichuan (SC); 17. Tibet (T); 18. Yunnan (YN); and 19. Zhejiang (ZJ). The partial sequences of RdRp gene (327-bp) of CoVs detected in Rhinolophus bats were aligned with those of published represenative CoV strains. The tree was constructed by the maximum-likelihood method with bootstrap values determined with 1000 replicates. The scale bar indicates the estimated number of substitutions per 10 nucleotides. Filled triangles indicate the CoVs published previously by our lab (KU343197, KP876536, KP876544, MF094687, KP876546, KY417143, FJ588686) [15.18.40.41], filled diamonds indicate CoVs detected in this study. Putative novel alphaCoVs are labeled in green. BtCoV/Rh/YN2012 detected in Guangdong and Yunnan province in this study are in bold. FIPV, Feline infectious peritonitis virus; PEDV, procine epidemic diarrhea virus; MHV, mouse hepatitis virus. Other abbreviations are defined as those in the text. Numbers in parentheses indicate numbers of sequences sharing >97% identity.
I doubt that RaBtCoV/4991’s place in that tree was determined based solely on a 370-bp fragment, so I would think that by early 2019, Shi Zhengli’s group would have sequenced its full genome.
Intriguingly, both pangolin-2017 and pangolin-2019 genomes are also very close in this stretch of the RdRp gene, and CoV2 and pangolin-2019 share a few common mutations not found in RaTG13:

But let’s put this topic aside for now and get back to the story of Shi Zhengli’s famous 2013 Nature paper.