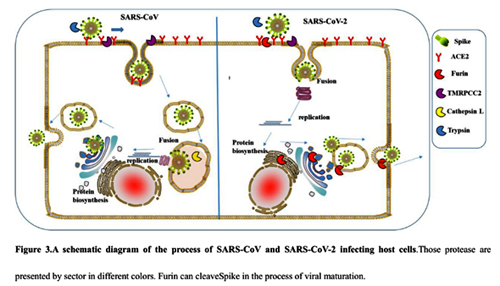
Did the SARS-CoV-2 virus arise from a bat coronavirus research program in a Chinese laboratory? Very possibly.
by Milton Leitenberg
Bulletin of the Atomic Scientists
June 4, 2020
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.
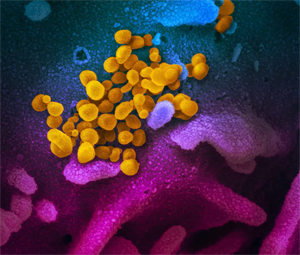
This scanning electron microscope image shows SARS-CoV-2 in yellow. This scanning electron microscope image shows SARS-CoV-2 in yellow. (Photo credit: Credit: NIAID-RML)
On May 15, the Bulletin of the Atomic Scientists published a short commentary titled, “Let evidence, not talk radio, determine whether the outbreak started in a lab,” by Ali Nouri, a biologist and president of the Federation of American Scientists. “The outbreak” referred to the pandemic of SARS-CoV-2 now circling the globe. It is a thin commentary, and it is puzzling why the Bulletin thought it desirable to publish it at all. Only two weeks earlier the journal had published a reasoned and competent appraisal by Kings College London biosecurity expert Filippa Lentzos titled, “Natural spillover or research lab leak? Why a credible investigation is needed to determine the origin of the coronavirus pandemic.”
The Nouri article very correctly pilloried the statements by President Donald Trump, Secretary of State Mike Pompeo, presidential legal advisor Rudy Giuliani, and radio personality Rush Limbaugh. These are as notorious a gang of four fabricators as will ever likely be recorded in American history. They were ably assisted by Fox News, which the Nouri critique also mentions. Nouri ended his commentary with these lines: “Our leaders ought to … take steps to prevent the next pandemic, instead of diverting our attention to unsupported sensationalist theories spread by cable TV and talk radio.”
Perhaps the most damaging blows to efforts to obtain a certain answer as to the origin of the SARS-CoV-2 “outbreak” have been the pronouncements by Trump, Pompeo, and their echo chambers. But they and their remarks are not the measure by which the question of the possibility that a laboratory escape began the pandemic should be examined. Trump’s diversionary ranting comes from a president who did nothing for two months in the face of an oncoming lethal pandemic, actively denied and denigrated intelligence warnings of the imminent danger, and said that SARS-CoV-2 would “just go away … like a miracle” and that “within a couple of days is going to be down close to zero.” All this has been widely and thoroughly chronicled.1
But long before Trump, Pompeo and Co. sought a Chinese scapegoat for the president’s gross and willful incompetence, researchers understood that the possibility of laboratory escape of the pathogen was a plausible, if unproven, possibility. It is most definitely not “a conspiracy theory.”
The circumstantial evidence for a lab escape.
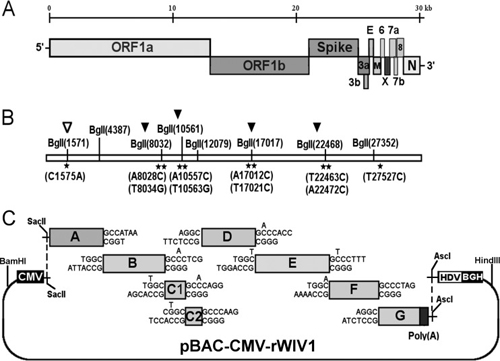
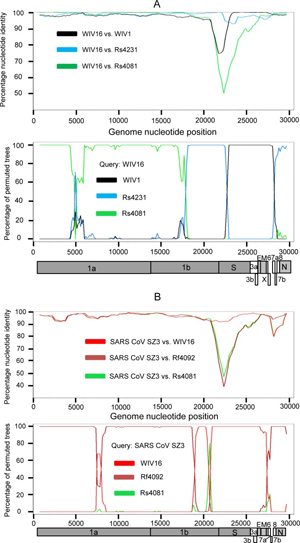
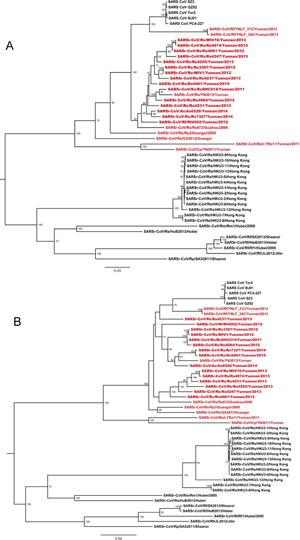
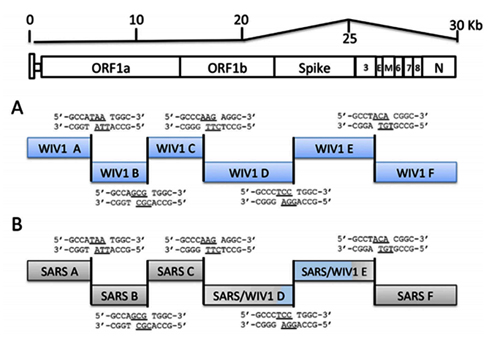
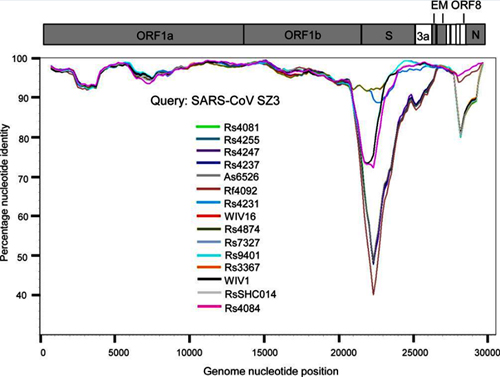
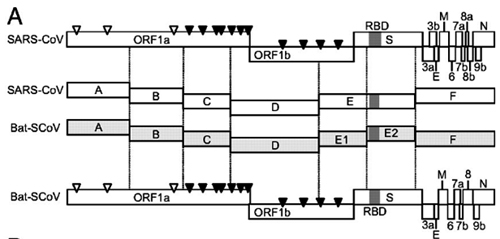
By way of introduction, there are two virology institutes in Wuhan to consider, not one: The Wuhan Center for Disease Control and Prevention (WHCDC) and the Wuhan Institute of Virology (WIV). Both have conducted large projects on novel bat viruses and maintained large research collections of novel bat viruses, and at least the WIV possessed the virus that is the most closely related known virus in the world to the outbreak virus, bat virus RaTG13. This virus was isolated in 2013 and had its genome published on January 23, 2020. Seven more years of bat coronavirus collection followed the 2013 RaTG13 isolation.
One component of the novel-bat-virus project at the Wuhan Institute of Virology involved infection of laboratory animals with bat viruses. Therefore, the possibility of a lab accident includes scenarios with direct transmission of a bat virus to a lab worker, scenarios with transmission of a bat virus to a laboratory animal and then to a lab worker, and scenarios involving improper disposal of laboratory animals or laboratory waste.

A May 20, 2020, photo of the Wuhan Institute of Virology in Wuhan, where research on bat coronaviruses was conducted. (Photo by Kyodo News via Getty Images)
Documentary evidence indicates that the novel-bat-virus projects at Wuhan CDC and the Wuhan Institute of Virology used personal protective equipment and biosafety standards that would pose high risk of accidental infection of a lab worker upon contact with a virus having the transmission properties of the outbreak virus.
In assessing the possibility of a lab accident, one must take into consideration each of the following eight elements of circumstantial evidence:
1. Official Chinese government recognition early in the SARS-CoV-2 outbreak of biosafety inadequacies in China’s high containment facilities.
In February 2020, several weeks after the outbreak of the disease in Wuhan, China’s President Xi Jinping stressed the need to ensure “biosafety and biosecurity of the country.”2 This was followed immediately by a China Ministry of Science & Technology announcement of new guidelines for laboratories, especially in handling viruses.3 Almost at the same time, the Chinese newspaper Global Times published an article on “chronic inadequate management issues at laboratories, including problems of biological wastes.”4
A PBS NewHour presentation on May 22, 2020 provided the following information:
On January 1, Wuhan Institute of Virology’s director general, Yanyi Wang, messaged her colleagues, saying the National Health Commission told her the lab’s COVID-19 data shall not be published on social media and shall not be disclosed to the media. And on January 3, the commission sent this document, never posted online, but saved by researchers, telling labs to destroy COVID-19 samples or send them to the depository institutions designated by the state. Late Friday [May 16, 2020] the Chinese government admitted to the destruction … but said it was for public safety.
The Chinese government explanation for the destruction of SARS-CoV-2 samples has no scientific credibility. For purposes of “public safety” any samples would surely be stored and studied, exactly as with the ones that were isolated from patients, and their RNA genomes decoded and published.
2. Recognition by Zhengli Shi, a renowned scientist who leads a research team at the Wuhan Institute of Virology, that a laboratory escape was a possibility.
Shi took the possibility of a laboratory escape perfectly seriously. Jonna Mazat of the University of California-Davis, a collaborator with Dr Shi, told Josh Rogin of the Washington Post, “Absolutely, accidents can happen.” In an interview with Scientific American, Shi admitted that her very first thought was “If coronaviruses were the culprit, she remembers thinking ‘Could they have come from our lab?’”
Meanwhile she frantically went through her own lab’s records from the past few years to check for any mishandling of experimental materials, especially during disposal. She breathed a sigh of relief when the results came back: none of the sequences matched those of the viruses her team had sampled from bat caves. ‘That really took a load off my mind,’ she says. ‘I had not slept a wink for days.’
3. Questions surrounding Chinese government attribution of the Wuhan’s Huanan South China Seafood Market as the source of the SARS-CoV-2 virus.
Many China scholars noted that it was quite unusual for Chinese government authorities to identify Wuhan’s Huanan South China Seafood Market so quickly as the source of the outbreak. They thought this behavior so uncharacteristic that it raised suspicions in their minds. The authors of a newly published paper wrote that
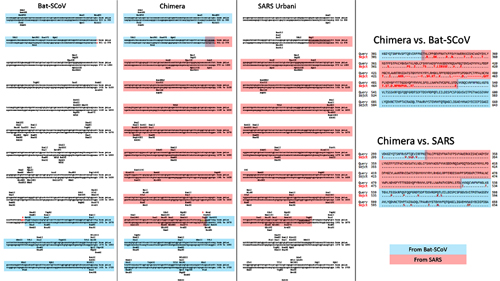
…we were surprised to find that SARS-CoV-2 resembles SARS-CoV in the late phase of the 2003 epidemic after SARS-CoV had developed several advantageous adaptations for human transmission. Our observations suggest that by the time SARS-CoV-2 was first detected in late 2019, it was already pre-adapted to human transmission to an extent similar to late epidemic SARS-CoV. However, no precursors or branches of evolution stemming from a less human-adapted SARS-CoV-2-like virus have been detected…. It would be curious if no precursor or branches of SARS-CoV-2 evolution are discovered in humans or animals….Even the possibility that a non-genetically-engineered precursor could have adapted to humans while being studied in a laboratory should be considered, regardless of how likely or unlikely.5
It is important to note that no intermediary host has yet been identified for the SARS-CoV-2 virus. The authors also noted that “[n]o animal sampling prior to the shutdown and sanitization [of the Wuhan fish market] was done.”

The question of whether the index case appeared in the Wuhan fish market appears to be moot in any case. Chinese researchers have published data showing that there were 41 cases of SARS-CoV-2 between December 1, 2019 and January 2, 2020. Fourteen of these had no contact with the Huanan seafood market, including the very first recorded case on December 1, 2019.6 And that supposes that the true index case was December 1, which is doubtful.
On May 26, the Chinese government scrapped the previous official story about the Wuhan fish market:
China’s top epidemiologist said Tuesday that testing of samples from a Wuhan food market, initially suspected as a path for the virus’s spread to humans, failed to show links between animals being sold there and the pathogen. Gao Fu, director of the Chinese Center for Disease Control and Prevention, said in comments carried in China state media.7
No SARS-CoV-2 isolates were detected in any of the animals or fish sold at the market, only in environmental samples, including sewage. Gao Fu added, “At first, we assumed the seafood market might have the virus, but now the market is more like a victim. The novel coronavirus had existed long before.”8
4. Suppression of information and individuals by Chinese authorities.
A publication by two Chinese university academics discussed both the WHCDC and the WIV and concluded that “the killer coronavirus probably originated from a laboratory in Wuhan”; the publication was removed from the internet by Chinese government officials. The paper had been posted on Research Gate but was blocked after 24 hours. After being placed on an archive file by internet users, it was again blocked after a week, and the two Chinese authors were pressured to retract the paper. However, it is still available on Web archives.9
The Chinese government closed the laboratory in Shanghai that first published the genome of COVID-19 on January 10, explaining that it had been shuttered for “rectification”; the closure happened on January 11. The government then permitted the same genome to be published by Shi on January 12.10 Chinese citizens who reported on the coronavirus were censured and, in some cases, “disappeared.”11 These have included businessman Fang Bin, ...
Fang Bin (Chinese: 方斌) is a Chinese businessman, citizen journalist and whistleblower who used Youtube and WeChat to broadcast images of Wuhan during the COVID-19 pandemic. He was arrested several times between February 1 and 9, 2020. He has been missing since his arrest on February 9, 2020...
On February 1, 2020, Fang released a new video showing the piling up of corpses at the back of a minivan in front of a Wuhan hospital. The video was shared on Twitter by Chinese journalist Jennifer Zeng. Fang was arrested on the same day, warned and eventually released during the night.
In February 2020, Fang and two other citizen journalists and whistleblowers, Chen Qiushi and Li Zehua, were arrested and went missing in Wuhan. Chen, a lawyer who arrived in Wuhan on January 23, disappeared first on February 6; one week prior, he had uploaded a video criticising the Chinese government's manipulation of information which ended with the sentence "I am not afraid to die! You think I am afraid of you communist party?". Li, another citizen journalist, went missing in late February, but reappeared in April posting a neutral, patriotic video, in contrast to his previous tone.
Two days before Chen's arrest, on February 4, the police came twice to Fang's apartment. He recorded the scene and refused to let them in without a warrant, worried that they had come in large numbers to arrest him (he counted at least four officers). Fang kept making videos from his apartment during the following days, criticising the government's propaganda and its choice to arrest Chen and Li Wenliang. On February 9, he released his last video: a 12-second clip showing a piece of paper with the sentence "resist all citizens, hand the power of the government back to the people" written on it. He has been missing ever since.
-- Fang Bin, by Wikipedia
lawyer Chen Qiushi,...
Chen Qiushi; born September 1985) is a Chinese lawyer, activist, and citizen journalist who covered the 2019–20 Hong Kong protests and the COVID-19 pandemic which included criticism of the government response. He was last heard from on 6 February 2020; as of July 2020, his whereabouts remain unknown. The Chinese government reportedly informed Chen's family and friends that he has been detained for the purpose of COVID-19 quarantine. Critics, including media freedom groups, have expressed skepticism about government motives, and have unsuccessfully called on the government to allow outside contact with Chen...
Chen began reporting on the COVID-19 pandemic in China, travelling to Hankou, Wuhan, on 23 or 24 January 2020, where he interviewed the locals and visited various hospitals including Huoshenshan Hospital, which was still under construction at the time. According to Chen, doctors were overworked and there were insufficient medical supplies, but prices of goods were otherwise stable. Chen published a video on 30 January showing the crowding in Wuhan hospitals, with many people lying in corridors. Unlike state media reporters who had hazmat suits, Chen appeared to have only goggles and a face mask to protect himself. Chen stated, "I am afraid. In front of me is disease. Behind me is China's legal and administrative power. But as long as I am alive I will speak about what I have seen and what I have heard. I am not afraid of dying. Why should I be afraid of you, Communist Party?— Chen Qiushi, 30 January 2020."
By early February 2020, while reporting about the coronavirus outbreak, Chen had 433,000 YouTube subscribers and 246,000 Twitter followers. Chen's supporters accused the Chinese government of censorship of the coronavirus outbreak. According to The Guardian, many pro-Chen comments on Sina Weibo were censored. Around 4 February, in the last video posted by Chen before his subsequent disappearance, Chen interviewed Wuhan resident "A Ming". A Ming stated his father had probably contracted coronavirus during a health check-up in the beginning of January, when there were no safety precautions; A Ming's father had subsequently died from the virus. During the video Chen stated "many people are worried I will be detained"...
Chen disappeared on 6 February 2020, at some point after informing his family of an intention to report on a temporary hospital. His friends were unable to contact him after 7 pm UTC+8 on 6 February. His mother, and friend Xu Xiaodong, have both stated that on 7 February, they received news from authorities that Chen had been detained at an undeclared time and place and held in an unknown location for the purpose of quarantine...
Around March 2020 it was reported that Li Zehua, a citizen journalist in part inspired by Chen, had also disappeared...
On 23 March, the Chinese Ambassador to the US, Cui Tiankai, stated he'd never heard of Chen... Li Zehua resurfaced in April 2020, stating he had been released on 28 March from a quarantine.-- Chen Qiushi, by Wikipedia
former state TV reporter Li Zehua ...
Li Zehua... is a Chinese citizen journalist, rapper and YouTuber... After graduating from Communication University of China, he joined China Central Television (CCTV) as a television presenter in 2016.
During the COVID-19 pandemic in China, he resigned from CCTV and found a way to get into Wuhan, hoping to trace disappeared journalist Chen Qiushi. With the help of locals, he was able to get a car and find a place to stay. In the following days, he used a vlog to report on the pandemic in Wuhan. He disappeared on 26 February 2020, presumed detained by officers from state security. Parts of his chase with the Wuhan authorities was caught on video and uploaded to YouTube. It was reported that Li Zehua returned to the hotel on 28 February. However, other reports stated that no one has heard from Li since his 26 February 2020 disappearance.
On 22 April 2020, Li posted a video on Youtube, Twitter, and Weibo, and uploaded the English subtitle to YouTube in the following days. According to Li, he was escorted on 26 February to the police station and was under investigation for disrupting public order. Additionally, police detained and quarantined him, citing his visits to sensitive epidemic areas. Li's quarantine was at first in Wuhan, and later moved to his hometown. Li stated that he had been treated well by the police during the detention, and that he had been released on 28 March...
At the end of his April 22 post, he quoted the Book of Documents aphorism "the mind of man is restless, prone (to err); its affinity to what is right is small. Be discriminating, be uniform (in the pursuit of what is right), that you may sincerely hold fast the Mean."... and gave his own interpretation in English:
The will of the people is unpredictable, the heart of Tao (the essence of cosmos) is fathomless. In order to make the will of the people consistent with the heart of Tao and reach the state of Unity of people and cosmos [zh], only the way is concentrate all energy on cultivation of the good nature of the heart, do not act in extreme, do not change faith, do not be fickle, uphold the doctrine of the golden mean of Confucian orthodoxy.
-- Li Zehua, by Wikipedia
and, most recently, Zhang Zhan, a lawyer.
A "citizen journalist" who has reported on the coronavirus emergency in Wuhan (Hubei), the epicenter of the pandemic, has been detained in a prison in Shanghai, the city where she resides, since February.
Zhang Zhan had disappeared on May 13, after live YouTube streaming from Hankou train station square. According to some of her friends, she was arrested on May 15 for "disturbing social stability and creating problems of public order".
In her stories, posted on YouTube, Twitter and other social media, the 37-year-old often attacked the Chinese government, whom she reputed at fault in having inadequately managed the crisis and depriving the Chinese people of their fundamental rights. In a February 16 Twitter post, Zhang accuses the authorities of lying about the actual number of Covid-19 victims to maintain stability.
During her stay in Wuhan, she also provided legal assistance to Yang Min, a woman who had asked for justice for her daughter who died of coronavirus, and therefore was placed under house arrest. Zhang had been arrested as early as September in Shanghai: she spent 60 days in prison for demonstrating in favor of Hong Kong's pro-democracy demonstrators.
Three other "citizen journalists" disappeared in Wuhan in February. Li Zehua, who had talked about the crematoriums of the city open 19 hours a day, reappeared on April 22 after a period under arrest. However, there is no news of Fang Bin and Chen Qiushi...
Since mid-March there has been no news of Ai Fen, the Wuhan doctor who raised the alarm on the disease. In the same days, Ren Zhiqiang, a billionaire who was already a member of the Chinese Communist Party (CCP), was also targeted for referring to President Xi Jinping as a "power hungry clown".
Since February there has been no news even of Xu Zhangrun and He Weifang. The two intellectuals had criticized the regime, arguing that the lack of press freedom has favored the spread of the coronavirus. A Shandong university student, Zhang Wenbin, disappeared on March 30 after posting a video asking for Xi to resign.
Chen Zhaozhi, a retired professor at Beijing University of Science and Technology, was imprisoned in Beijing on April 14. He said that Covid-19 is not a "Chinese virus", but a "Chinese Communist Party virus", linking the origin and spread of the lung disease to the regime.
-- Zhang Zhan, a 'journalist citizen' who disappeared in Wuhan, arrested, by AsiaNews.it
They are reportedly being held in extrajudicial detention centers for speaking out about China’s response to the pandemic. They are usually accused of “picking quarrels and provoking trouble.”12
Another aspect of Chinese government secrecy involved in the SARS-CoV-2 pandemic relates to official reporting by Chinese government officials on the severity of the outbreak in China and on levels of mortality. The number of cases and deaths are suspected of being undercounted by at least an order of magnitude, and possibly two, meaning that the reported figures could be as little as one percent of the actual totals. In the last week of April 2020, Caixin, one of the most reliable publications in China, reported that a serological study had been carried out in Wuhan on 11,000 inhabitants. Extrapolating from its results, which showed that five to six percent of the sample of 11,000 persons carried antibodies for SARS-CoV-2, Caixin estimated that 500,000 people in the city had been infected, or 10 times the level of official Chinese government reporting. The publication was quickly deleted by Chinese government censors.13
The Chinese government has also attempted to obscure the origins of the pandemic with disinformation. On March 13, Chinese Foreign Ministry spokesperson Zhao Lijian suggested that the United States might have introduced the coronavirus to Wuhan.14
The US National Institutes of Health (NIH) funded bat-coronavirus research in the Wuhan Institute of Virology in China to the tune of US $3.7 million, a recent article in the British newspaper Daily Mail revealed.
Back in October 2014, the US government had placed a federal moratorium on gain-of-function (GOF) research -– altering natural pathogens to make them more deadly and infectious -– as a result of rising fears about a possible pandemic caused by an accidental or deliberate release of these genetically engineered monster germs.
This was in part due to lab accidents at the US Centers for Disease Control and Prevention (CDC) in July 2014 that raised questions about biosafety at US high-containment labs.
At that time, the CDC had closed two labs and halted some biological shipments in the wake of several incidents in which highly pathogenic microbes were mishandled by US government laboratories: an accidental shipment of live anthrax, the discovery of forgotten live smallpox samples and a newly revealed incident in which a dangerous influenza strain was accidentally shipped from the CDC to another lab.
A CDC internal report described how scientists failed to follow proper procedures to ensure samples were inactivated before they left the lab, and also found “multiple other problems” with operating procedures in the anthrax lab.
As such in October 2014, because of public health concerns, the US government banned all federal funding on efforts to weaponize three viruses –- influenza, Middle East respiratory syndrome (MERS) and severe acute respiratory syndrome (SARS).
In the face of a moratorium in the US, Dr Anthony Fauci –- the director of the National Institute of Allergy and Infectious Diseases (NIAID) and currently the leading doctor in the US Coronavirus Task Force –- outsourced in 2015 the GOF research to China’s Wuhan lab and licensed the lab to continue receiving US government funding...
Nonetheless, it is unclear what the legal ramifications would be if the virus was indeed leaked from a Chinese lab, but as a result of a research project that was outsourced and funded by the US government.
Also, if there was a government ban in 2014 on federal funding being used for GOF research, what are the federal compliance and ethical issues surrounding the fact that the NIH still gave federal funding instead of private funding to the Wuhan lab to continue the experiments?
Moreover, could some strains of the coronavirus have originated in U.S. labs, given the fact the US government lifted the ban in December 2017 on GOF research without resolving lab-safety issues?
-- Why US outsourced bat virus research to Wuhan: US-funded $3.7 million project approved by Trump's Covid-19 guru Dr Anthony Fauci in 2015 after US ban imposed on 'monster-germ' research, by Christina Lin
A month later, Zhao Lijian again posted Russian coronavirus and biowarfare-related disinformation, this time followed by online posts from Chinese ambassadors in 13 countries spread across the world.15
Meanwhile Chinese government officials have promoted baseless conspiracy theories about the virus’s origins, claiming that the United States military brought it to Wuhan. Zhao Lijian, a spokesperson for the Chinese Ministry of Foreign Affairs, retweeted one such article from the fringe conspiracy website Global Research.
-- Analyzing China’s Coronavirus Propaganda Messaging in Europe, by Matt Schrader
This was unprecedented diplomatic behavior for China, but not an accident. It was a concerted, deliberate, and preposterous disinformation campaign, repeated in May by CGTN, the China Global Television Network, which reposted the disinformation to the social media sites Weibo, Facebook, and Twitter.16 The history of Soviet and then Russian government biowarfare disinformation suggests that a country spreading such disinformation has or may have something to hide.17
You know Ralph, I think the public has not been in on this debate. It has been a secret debate with the NIH and other people who funded it. I think COVID-19 is a product of this. And I know Trump wants to call it the China virus. Well, the money that went into the creation and the genetic engineering of these coronaviruses in Wuhan was supported by the NIH and the USAID. So why wouldn't it be the NIH virus, or the USAID virus?
-- Interview with Andrew Kimball on the Ralph Nader Radio Show, Ralph Nader Radio Hour Episode 332
5. Laboratory accidents and the escape of highly dangerous pathogens from laboratories are frequent occurrences worldwide.
The accidental infection of researchers in the highest containment biosafety facilities—labelled BSL-2, BSL-3 and BSL-4—occurs worldwide, as do accidental releases by other means. In an excellent review published in February 2019, Lynn Klotz of the Center for Arms Control and Non-Proliferation noted that three releases of Ebola and Marburg viruses from BSL-4 and lower-containment facilities in the United States had occurred due to incomplete inactivation of cultures. Releases via infection of researchers took place in the highest containment facilities in the United States—at the Centers for Disease Control and Prevention (CDC) in Atlanta and at the US Army Medical Research Institute of Infectious Diseases (USAMRIID)—but in all cases only the researcher became ill, and there was no further transmission of the pathogen.
“In an analysis circulated at the 2017 meeting for the Biological Weapons Convention, a conservative estimate shows that the probability is about 20 percent for a release of a mammalian-airborne-transmissible, highly pathogenic avian influenza virus into the community from at least one of 10 labs over a 10-year period of developing and researching this type of pathogen,” Klotz wrote. “This percentage was calculated from FSAP [US Federal Select Agent Program] data for the years 2004 through 2010. Analysis of the FOIA NIH (National Institutes of Health) data gives a much higher release probability—that is, a factor five to 10 times higher, based on a smaller number of incident reports.”18
Between 2009 and 2015, the FSAP recorded 749 incidents in seven categories—not solely releases or researcher infections—from 276 facilities. In addition, Klotz recorded 11 confirmed releases of select agents that resulted in a laboratory-acquired infection in roughly 280 specifically approved laboratories in the United States between 2003 and 2017, a rate of just under one per year.19 A second publication in the Bulletin that covered closely-related subject matter and a personal communication from its author suggested that federally reported cases involving select agents were likely to be substantially undercounted:20
There is a fundamental problem of using the defined select agents as a surrogate for potential pandemic agent releases from research labs. The vast majority of ‘classical BW agents’ that initially defined select agents in the US were selected specifically to be NOT capable of sustained transmission so as to better define the military tactical limits of a military employment and because the establishment of progressive transmission was considered unpredictable and possibly counterproductive in military operations, at least on the US side of offensive development in the 1940s-1960s.
As my historical review of lab escapes that resulted in pandemics or wide area epidemics published in the BAS found, most pandemic, continental or large scale community outbreaks originating from lab escapes came from civilian labs working with public or veterinary pathogens of non-military interest.
It takes only one superspreading graduate student or maintenance worker to start a pandemic.
It is known that a very large percentage of the individuals infected with the SARS-CoV-2 virus show no symptoms and do not become clinically ill, which would facilitate an unrecognized infection of one or more laboratory researchers.
6. There have been laboratory accidents and escapes of highly dangerous pathogens in China in general and biosafety issues at the Wuhan Institute of Virology in particular.
After the SARS epidemic in 2002-2003, which originated naturally in China and which China initially kept secret, work on the coronavirus pathogen that was responsible for the outbreak was undertaken in laboratories around the world. This research led to six cases of infection in laboratory workers: four in the National Institute of Virology in Beijing and one each in laboratories in Singapore and Taiwan.
The laboratory-acquired infections of lab workers in Beijing led to short-lived outbreaks of SARS in the Beijing region in 2004.21
A second case of infected researchers in China resulted in brief outbreaks of disease in early December 2019. An outbreak of brucellosis began in an agricultural laboratory in Lanzhou (Gansu Province, central China) and spread to China’s premier bird flu laboratory in Harbin (Heilongjiang Province, northeast China). It was linked to index cases involving graduate students who were exposed while conducting research and included at least 96 people.22
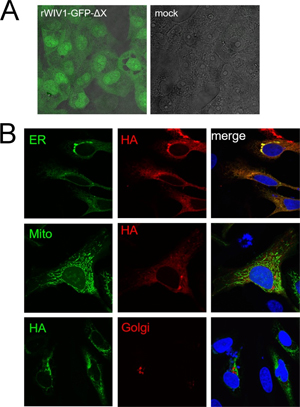
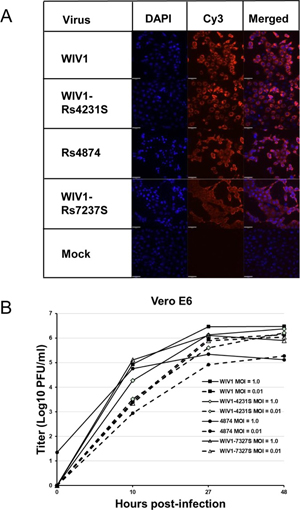
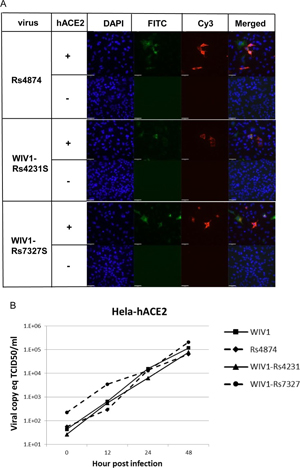
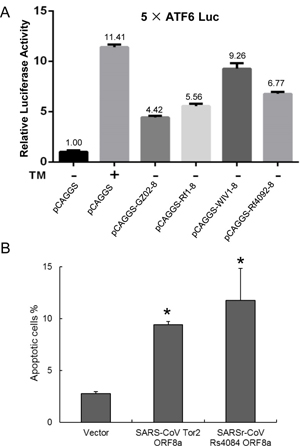
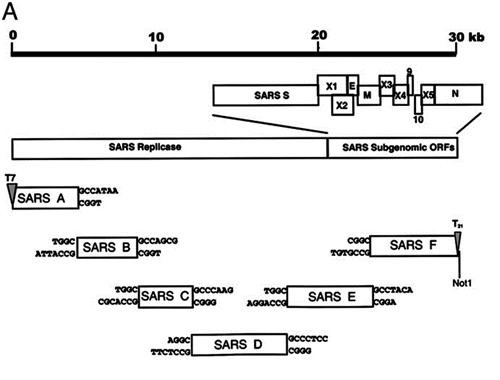
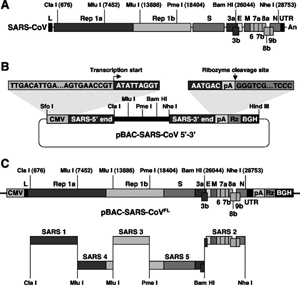
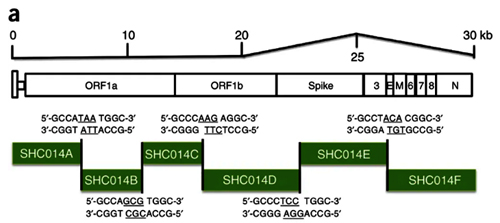
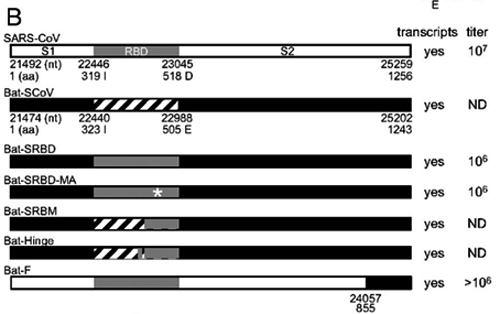
7. Under what biosafety conditions was bat coronavirus research carried out at the Wuhan Institute of Virology?
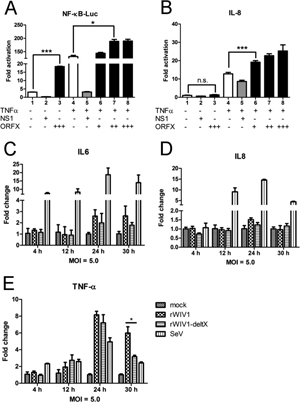
Most work—including all published work using live bat coronaviruses that were not SARS-CoV and MERS-CoV—was conducted under BSL-2 conditions.23 This was consistent with both WHO and CDC recommendations.24 BSL-2 provides only minimal protection against infection of laboratory researchers, and these regulations were almost certainly too lenient for working with bat coronaviruses. All such work should have been carried out under BSL-3 conditions. However, extremely high-risk gain of function (GoF) studies with bat SARS-related coronaviruses were carried out at BSL-3 or BSL-4. Statements made by various commentators claiming that the WIV worked only with RNA isolates and not with live viruses are untrue (as discussed in further detail in a following section).