Re: U.S. government gave $3.7 million grant to Wuhan lab at
Part 1 of 2
Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus
by Ben Hu, Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – original draft,#1 Lei-Ping Zeng, Investigation, Methodology, Xing-Lou Yang, Investigation, Resources, Xing-Yi Ge, Formal analysis, Resources, Wei Zhang, Investigation, Bei Li, Investigation, Jia-Zheng Xie, Investigation, Xu-Rui Shen, Investigation, Yun-Zhi Zhang, Resources, Ning Wang, Investigation,1 Dong-Sheng Luo, Investigation, Resources,1 Xiao-Shuang Zheng, Investigation, Mei-Niang Wang, Resources,1 Peter Daszak, Funding acquisition, Writing – review & editing, Lin-Fa Wang, Conceptualization, Funding acquisition, Writing – review & editing, Jie Cui, Conceptualization, Formal analysis, Funding acquisition, Software, Writing – review & editing, and Zheng-Li Shi, Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – review & editing
Christian Drosten, Editor
Copyright © 2017 Hu et al
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
Abstract
A large number of SARS-related coronaviruses (SARSr-CoV) have been detected in horseshoe bats since 2005 in different areas of China. However, these bat SARSr-CoVs show sequence differences from SARS coronavirus (SARS-CoV) in different genes (S, ORF8, ORF3, etc) and are considered unlikely to represent the direct progenitor of SARS-CoV. Herein, we report the findings of our 5-year surveillance of SARSr-CoVs in a cave inhabited by multiple species of horseshoe bats in Yunnan Province, China. The full-length genomes of 11 newly discovered SARSr-CoV strains, together with our previous findings, reveals that the SARSr-CoVs circulating in this single location are highly diverse in the S gene, ORF3 and ORF8. Importantly, strains with high genetic similarity to SARS-CoV in the hypervariable N-terminal domain (NTD) and receptor-binding domain (RBD) of the S1 gene, the ORF3 and ORF8 region, respectively, were all discovered in this cave. In addition, we report the first discovery of bat SARSr-CoVs highly similar to human SARS-CoV in ORF3b and in the split ORF8a and 8b. Moreover, SARSr-CoV strains from this cave were more closely related to SARS-CoV in the non-structural protein genes ORF1a and 1b compared with those detected elsewhere. Recombination analysis shows evidence of frequent recombination events within the S gene and around the ORF8 between these SARSr-CoVs. We hypothesize that the direct progenitor of SARS-CoV may have originated after sequential recombination events between the precursors of these SARSr-CoVs. Cell entry studies demonstrated that three newly identified SARSr-CoVs with different S protein sequences are all able to use human ACE2 as the receptor, further exhibiting the close relationship between strains in this cave and SARS-CoV. This work provides new insights into the origin and evolution of SARS-CoV and highlights the necessity of preparedness for future emergence of SARS-like diseases.
Author summary
Increasing evidence has been gathered to support the bat origin of SARS coronavirus (SARS-CoV) in the past decade. However, none of the currently known bat SARSr-CoVs is thought to be the direct ancestor of SARS-CoV. Herein, we report the identification of a diverse group of bat SARSr-CoVs in a single cave in Yunnan, China. Importantly, all of the building blocks of SARS-CoV genome, including the highly variable S gene, ORF8 and ORF3, could be found in the genomes of different SARSr-CoV strains from this single location. Based on the analysis of full-length genome sequences of the newly identified bat SARSr-CoVs, we speculate that the direct ancestor of SARS-CoV may have arisen from sequential recombination events between the precursors of these bat SARSr-CoVs prior to spillover to an intermediate host. In addition, we found bat SARSr-CoV strains with different S proteins that can all use the receptor of SARS-CoV in humans (ACE2) for cell entry, suggesting diverse SARSr-CoVs capable of direct transmission to humans are circulating in bats in this cave. Our current study therefore offers a clearer picture on the evolutionary origin of SARS-CoV and highlights the risk of future emergence of SARS-like diseases.
Introduction
Severe Acute Respiratory Syndrome (SARS) is a severe emerging viral disease with high fatality characterized by fever, headache and severe respiratory symptoms including cough, dyspnea and pneumonia [1]. Due to its high transmissibility among humans, after its first emergence in southern China in late 2002, it rapidly led to a global pandemic in 2003 and was marked as one of the most significant public health threats in the 21st century [2,3]. The causative agent, SARS coronavirus (SARS-CoV), has been previously assigned to group 2b CoV and is now a member of the lineage B of genus Betacoronavirus in the family Coronaviridae [4]. It shares similar genome organization with other coronaviruses, but exhibits a unique genomic structure which includes a number of specific accessory genes, including ORF3a, 3b, ORF6, ORF7a, 7b, ORF8a, 8b and 9b [5,6].
Masked palm civets (Paguma larvata) were initially hypothesized to be the animal origin of SARS-CoV [7,8]. However, since a large number of genetically diverse SARS-related coronaviruses (SARSr-CoV) have been detected in multiple species of horseshoe bats (genus Rhinolophus) from different areas of China and Europe in the aftermath of SARS, it is prevailingly considered that SARS-CoV originated in horseshoe bats with civets acting as the intermediate amplifying and transmitting host [9–16]. Recently we have reported four novel SARSr-CoVs from Chinese horseshoe bats that shared much higher genomic sequence similarity to the epidemic strains, particularly in their S gene, of which two strains (termed WIV1 and WIV16) have been successfully cultured in vitro [17,18]. These newly identified SARSr-CoVs have been demonstrated to use the same cellular receptor (angiotensin converting enzyme-2 [ACE-2]) as SARS-CoV does and replicate efficiently in primary human airway cells [17–19].
Despite the cumulative evidence for the emergence of SARS-CoV from bats, all bat SARSr-CoVs described so far are clearly distinct from SARS-CoV in the S gene and/or one or more accessory genes such as ORF3 and ORF8, suggesting they are likely not the direct ancestor of SARS-CoV. Thus a critical gap remains in our understanding of how and where SARS-CoV originated from bat reservoirs. Previously, we reported a number of bat SARSr-CoVs with diverse S protein sequences from a single cave in Yunnan Province, including the four strains mentioned above most closely related to SARS-CoV [17,18]. Here we report the latest results of our 5-year longitudinal surveillance of bat SARSr-CoVs in this single location and systematic evolutionary analysis using full-length genome sequences of 15 SARSr-CoV strains (11 novel ones and 4 from previous studies). Efficiency of human ACE2 usage and the functions of accessory genes ORF8 and 8a were also evaluated for some of the newly identified strains.
Results
Continued circulation of diverse SARSr-CoVs in bats from a single location
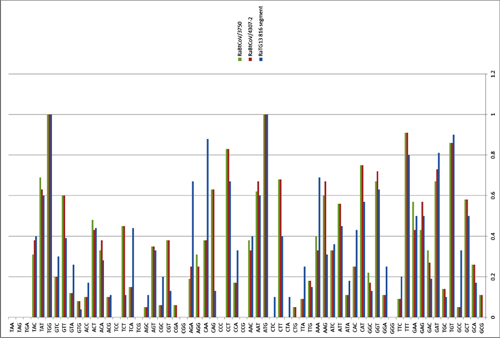
We have carried out a five-year longitudinal surveillance (April 2011 to October 2015) on SARSr-CoVs in bats from a single habitat in proximity to Kunming city, Yunnan province, China, which was mainly inhabited by horseshoe bats. A total of 602 alimentary specimens (anal swabs or feces) were collected and tested for the presence of CoVs by a Pan-CoV RT-PCR targeting the 440-nt RdRp fragment that is conserved among all known α- and β-CoVs [20]. In total, 84 samples tested positive for CoVs. Sequencing of the PCR amplicons revealed the presence of SARSr-CoVs in the majority (64/84) of the CoV-positive samples (Table 1). Host species identification by amplification of either Cytb or ND1 gene suggested that most (57/64) of the SARSr-CoV positive samples were from Rhinolophus sinicus, while the remaining 7 samples were from Rhinolophus ferrumequinum, Rhinolophus affinis and from Aselliscus stoliczkanus which belongs to the family Hipposideridae.
Based on the preliminary analysis of the partial RdRp sequences, all of the 64 bat SARSr-CoV sequences showed high similarity among themselves and with other reported bat SARSr-CoVs and SARS-CoVs from humans and civets. To understand the genetic diversity of these bat SARSr-CoVs, the most variable region of the SARSr-CoV S gene, corresponding to the receptor-binding domain (RBD) of SARS-CoV, were amplified and sequenced. Due to low viral load in some samples, RBD sequences were successfully amplified only from 49 samples. These RBD sequences displayed high genetic diversity and could be divided into two large clades, both of which included multiple genotypes. Clade 1 strains shared an identical size and higher amino acid (aa) sequence identity with SARS-CoV RBD, while clade 2 had a shorter size than SARS-CoV S due to two deletions (5 and 12–13 aa, respectively) (S1 Fig). Co-infections by two strains of different clades were detected in two samples, Rs3262 and Rs4087 (S1 Fig).
Genomic characterization of the novel SARSr-CoVs
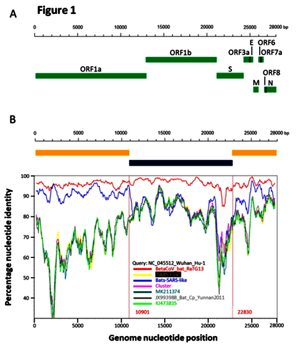
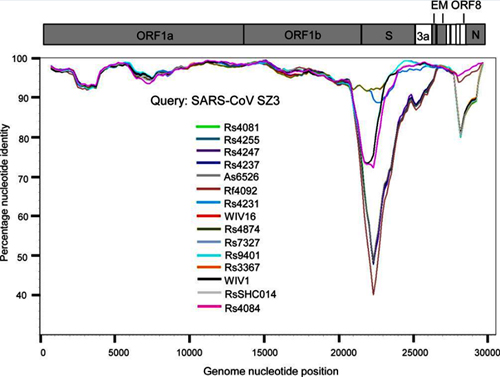
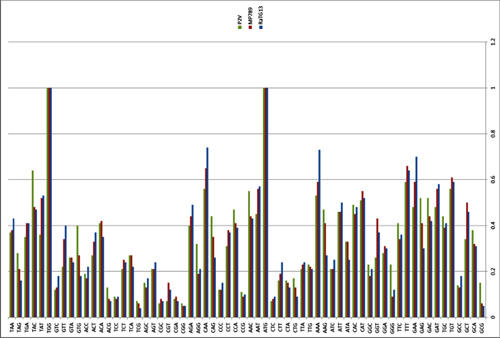
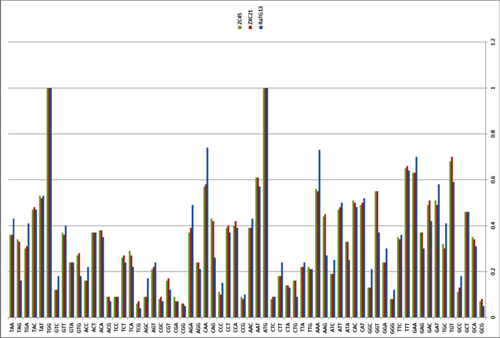
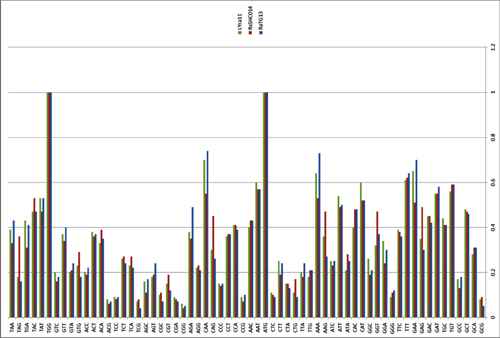
Based on the diversity of RBD sequences, 11 novel SARSr-CoV strains named by abbreviation of bat species and sample ID (Rs4081, Rs4084, Rs4231, Rs4237, Rs4247, Rs4255, Rs4874, Rs7327, Rs9401, Rf4092 and As6526) were selected for full-length genomic sequencing based on sample abundance, genotype of RBD as well as sampling time. For each RBD genotype and each time of sampling, at least one representative strain was selected. The genome size of these novel SARSr-CoVs ranged from 29694 to 30291 nucleotides (nt). This gave a total of 15 full-length genomes of bat SARSr-CoVs from this single location (13 from R.sinicus, and one each from R. ferrumequinum and A. stoliczkanus), including our previously reported strains, Rs3367, RsSHC014, WIV1 and WIV16 [17,18]. The genomes of all 15 SARSr-CoVs circulating in this single cave shared 92.0% to 99.9% nt sequence identity. The overall nt sequence identity between these SARSr-CoVs and human and civet SARS-CoVs is 93.2% to 96%, significantly higher than that observed for bat SARSr-CoVs reported from other locations in China (88–93%) [9,10,12,14,21,22]. The genome sequence similarity among the 15 SARSr-CoVs and SARS-CoV SZ3 strain was examined by Simplot analysis (Fig 1). The 15 SARSr-CoVs are highly conserved and share a uniformly high sequence similarity to SARS-CoV in the non-structural gene ORF1a (96.6% to 97.1% nt sequence identity, 98.0% to 98.3% aa sequence identity) and ORF1b (96.1% to 96.6% nt sequence identity, 99.0% to 99.4% aa sequence identity). In contrast, a considerable genetic diversity is shown in the S gene (corresponding to SZ3 genome position 21477 to 25244) and ORF8 (corresponding to SZ3 genome position 27764 to 28132) (Fig 1).
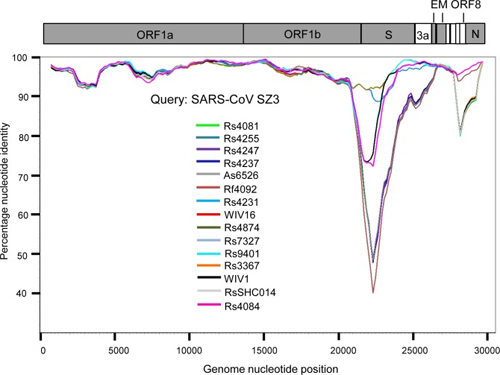
Fig 1. Similarity plot based on the full-length genome sequence of civet SARS CoV SZ3.
Full-length genome sequences of all SARSr-CoV detected in bats from the cave investigated in this study were used as reference sequences. The analysis was performed with the Kimura model, a window size of 1500 base pairs and a step size of 150 base pairs.
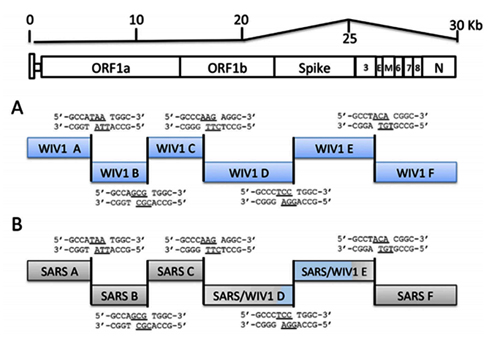
The 11 novel SARSr-CoVs identified from this single location generally shared similar genome organization with SARS-CoV and other bat SARSr-CoVs. In our previous study, we identified an additional ORF termed ORFx present between ORF6 and ORF7 in strain WIV1 and WIV16 [18,23]. In this study, ORFx was also found in the genomes of Rs7327 and Rs4874. Compared with that of WIV1 and WIV16, the length of ORFx in Rs7327 and Rs4874 was extended to 510 nt due to a deletion of 2 nt in a poly-T sequence that resulted in a shift of reading frame (Fig 2 and S2 Fig).
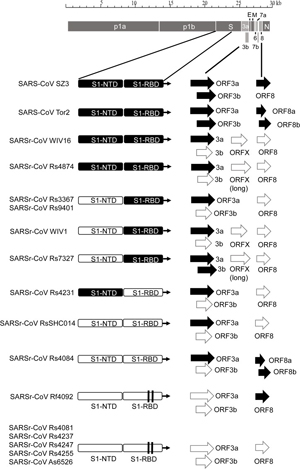
Fig 2. Schematic diagram illustrating the genomic regions or ORFs with most variation between different SARS-CoV and SARSr-CoV isolates.
Coding regions of the N-terminal domain (NTD) and receptor-binding domain (RBD) of the spike protein, ORF3a/b and ORF8 (8a/b) in bat SARSr-CoV genomes highly similar to those in SARS CoV genome are indicated with black boxes or arrows while the hollow boxes or arrows represent corresponding regions with less sequence similarity to those of SARS-CoV. The deletions in the RBD of some SARSr-CoVs are indicated by two vertical lines.
Co-circulation of different bat SARSr-CoVs with S, ORF8 and ORF3 sequences similar to those in SARS-CoV at a single location
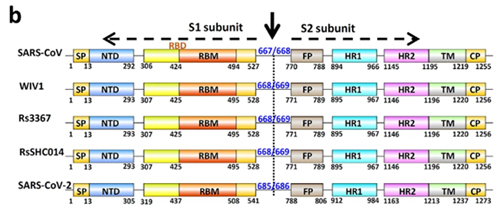
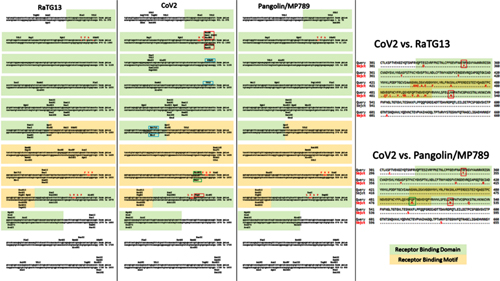
The primary difference between SARS-CoV and most bat SARSr-CoVs is located in S gene. The S protein is functionally divided into two subunits, denoted S1 and S2, which is responsible for receptor binding and cellular membrane fusion, respectively. S1 consists of two domains, the N-terminal domain (NTD) and C-terminal domain (CTD) which is also known as the RBD in SARS-CoV [24]. SARS-CoV and bat SARSr-CoVs share high sequence identity in the S2 region in contrast to the S1 region. Among the 15 SARSr-CoVs identified from bats in the surveyed cave, six strains with deletions in their RBD regions (Rs4081, Rs4237, Rs4247, Rs4255, Rf4092 and As6526) showed 78.2% to 80.2% aa sequence identity to SARS-CoV in the S protein, while the other nine strains without deletions were much more closely related to SARS-CoV, with 90.0% (Rs4084) to 97.2% (Rs4874) aa sequence identity. These nine SARSr-CoVs can be further divided into four genotypes according to their S1 sequences (Fig 2): RsSHC014/Rs4084 showed more genetic differences from SARS-CoV in both NTD and RBD regions; The RBD sequences of SARSr-CoV Rs7327, Rs9401 and previously reported WIV1/Rs3367 closely resembled that of SARS-CoV. However, they were distinct from SARS-CoV but similar to RsSHC014 in NTD. In contrast, we found a novel SARSr-CoV, termed Rs4231, which shared highly similar NTD, but not RBD sequence with SARS-CoV (Figs (Figs22 and and3).3). Its S protein showed 94.6% to 95% aa sequence identity to those of human and civet SARS-CoVs (S1 Table). Strains with both NTD and RBD highly homologous to those of SARS-CoV were also present in this cave. In addition to WIV16 which we described previously [18], Rs4874 was also found to have the S protein closest to SARS-CoV S (> 97% aa sequence identity) of all the bat SARSr-CoVs reported to date (Figs (Figs22 and and3).3). In addition to the SARSr-CoVs subjected to full-length genome sequencing, we also obtained the RBD and NTD sequences from other samples collected in this cave. The sequences with high identity to SARS-CoV RBD were amplified from 10 more R. sinicus samples. SARSr-CoVs with this genotype of RBD were detected in different seasons throughout the five years. Strains containing the NTD similar to SARS-CoV were only found in 2013 (S2 Table).

Fig 3. Amino acid sequence comparison of the S1 subunit (corresponding to aa 1–660 of the spike protein of SARS-CoV).
The receptor-binding domain (aa 318–510) of SARS-CoV and the homologous region of bat SARSr-CoVs are indicated by the red box. The key aa residues involved in the interaction with human ACE2 are numbered on top of the aligned sequences. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from civets in 2003. SARSr-CoV Rs 672 and YN2013 were identified from R. sinicus collected in Guizhou and Yunnan Province, respectively. SARSr-CoV Rf1 and JL2012 were identified from R. ferrumequinum collected in Hubei and Jilin Province, respectively. WIV1, WIV16, RsSHC014, Rs4081, Rs4084, Rs4231, Rs4237, Rs4247, Rs7327 and Rs4874 were identified from R.sinicus, and Rf4092 from R. ferrumequinum in the cave surveyed in this study.
ORF8 is another highly variable gene among different SARS-CoV and SARSr-CoV strains [25,26]. We aligned the ORF8 nt sequences of the representative SARSr-CoVs discovered in this surveillance with those of other SARSr-CoVs and SARS-CoVs (Fig 4). Though WIV16, WIV1, Rs4231 and RsSHC014 were genetically closer to SARS-CoV in S gene, they contained a single 366-nt ORF8 without the 29-nt deletion present in most human SARS-CoVs and showed only 47.1% to 51.0% nt sequence identity to human and civet SARS-CoVs. However, the ORF8 of strain Rf4092 from R. ferrumequinum exhibited high similarity to that of civet SARS-CoV. It possessed a single long ORF8 of the same length (369 nt) as that of civet SARS-CoV strain SZ3, with only 10 nt mutations and 3 aa mutations detected (Fig 4). Similar ORF8 sequences were also amplified from other 7 samples collected in the cave during 2011 to 2013, from both R. ferrumequinum and R. sinicus (S2 Table). The ORF8 of Rs4084 was highly similar to Rf4092’s but was split into two overlapping ORFs, ORF8a and ORF8b, due to a short 5-nt deletion (Figs (Figs22 and and4).4). The position of start codons and stop codons of the two ORFs were consistent with those in most human SARS-CoV strains. Excluding the 8-aa insertion, Rs4084 and SARS-CoV strain BJ01 displayed identical aa sequence of ORF8a, and only three different aa residues were observed between their ORF8b (Fig 4). To our knowledge, Rs4084 was the first bat SARSr-CoV reported that resembled the late human SARS-CoVs in both ORF8 gene organization and sequence.

Fig 4. Alignment of nucleotide sequences of ORF8 or ORF8a/8b.
The start codons and stop codons of ORF8, 8a and 8b are marked with black boxes and the forward and reverse arrows, respectively. The deletion responsible for the split ORF8a and 8b in human SARS-CoV BJ01, Tor2 and bat SARSr-CoV Rs4084 is marked with red boxes. See the legend for Fig 3 for the origin of various sequences used in this alignment.
Another key difference between SARS-CoV and bat SARSr-CoV genomes is the ORF3 coding region [10,17,21]. We analyzed the ORF3a sequences amplified from 42 samples and found that most of the SARSr-CoVs closely related to SARS-CoV in the S gene shared higher ORF3a sequence similarity (96.4% to 98.9% aa identity) with SARS-CoV (S3 Fig and S2 Table). The ORF3b of SARS CoV, sharing a large part of its coding sequence with the ORF3a, encodes a 154-aa protein [27], but it is truncated to different extents at the C-terminal in previously described bat SARSr-CoVs including WIV1 and WIV16 (S4 Fig). In the current study, we identified a non-truncated ORF3b for the first time (Rs7327), which maintained the nuclear localization signal at its C-terminal. Moreover, it shared 98.1% aa sequence identity with SARS-CoV strain Tor2 with only three aa substitutions (S4 Fig). Thus, Rs7327 is the bat SARSr-CoV most similar to SARS-CoV in the ORF3 region known to date.
Recombination analysis
The full-length genome sequences of all 15 SARSr-CoVs from the surveyed cave were screened for evidence of potential recombination events. Both similarity plot and bootscan analyses revealed frequent recombination events among these SARSr-CoV strains. It was suggested that WIV16, the closest progenitor of human SARS-CoV known to date [18], was likely to be a recombinant strain from three SARSr-CoVs harbored by bats in the same cave, namely WIV1, Rs4231 and Rs4081, with strong P value (<10−30). Breakpoints were identified at genome positions nt 18391, 22615 and 28160 (Fig 5A). In the genomic region between nt 22615 and 28160, which contained the region encoding the RBD and the S2 subunit of the S protein, WIV16 was highly similar to WIV1, sharing 99% sequence identity. In contrast, in the region between nt 18391 and 22615, which covered a part of ORF1b and the region encoding the NTD of the S gene, WIV16 showed substantially closer relationship to Rs4231. Meanwhile, the ORF1ab sequences upstream from nt 18391 of WIV16 displayed the highest genetic similarity (99.8% nt sequence identity) to that of Rs4081.
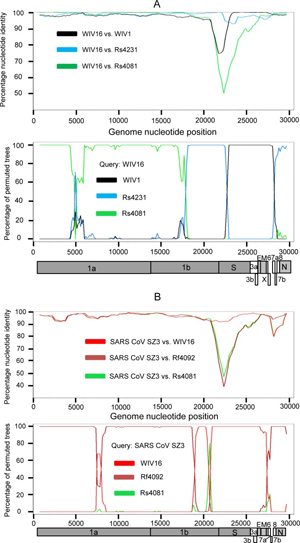
Fig 5. Detection of potential recombination events by similarity plot and boot scan analysis.
(A) Full-length genome sequence of SARSr-CoV WIV16 was used as query sequence and WIV1, Rs4231 and Rs4081 as reference sequences. (B) Full-length genome sequence of SARS-CoV SZ3 was used as query sequence and SARSr-CoV WIV16, Rf4092 and Rs4081 as reference sequences. All analyses were performed with a Kimura model, a window size of 1500 base pairs, and a step size of 150 base pairs. The gene map of query genome sequences are used to position breakpoints.
Evidence of recombination event was also detected in the genome of the novel SARSr-CoV Rs4084, which had a unique genome organization with split ORF8a and 8b. The previously reported strain RsSHC014 and the newly identified strain Rf4092 were suggested to be the major and minor parent of Rs4084, respectively (P value < 10−80). The breakpoint was located at nt 26796 (S5 Fig). In the region downstream of the breakpoint including ORF8, Rs4084 showed closet genetic relationship with Rf4092, sharing 98.9% nt sequence identity, while it shared the highest nt sequence identity (99.4%) with RsSHC014 in the majority of its genome upstream from the breakpoint.
When civet SARS-CoV SZ3 was used as the query sequence in similarity plot and bootscan analysis, evidence for recombination events was also detected (Fig 5B). In the region between the two breakpoints at the genome positions nt 21161 and nt 27766, including the S gene, closer genetic relationship between SZ3 and WIV16 was observed. However, from position nt 27766 towards the 3’ end of its genome, a notably close genetic relationship was observed between SZ3 and Rf4092 instead. Throughout the non-structural gene, moreover, SZ3 shared a similarly high sequence identity with WIV16 and Rf4092. It indicates that civet SARS-CoV was likely to be the descendent from a recombinant of the precursors of WIV16 and Rf4092, or that the SARSr-CoVs found in this cave, like WIV16 or Rf4092, may have been the descendants of the SARS-CoV lineage.
Phylogenetic analysis
Phylogenetic trees were constructed using the nt sequences of nonstructural protein gene ORF1a and ORF1b. Unlike the high genetic diversity in the S gene, nearly all SARSr-CoVs from the bat cave we surveyed were closely clustered, and showed closer phylogenetic relationship to SARS-CoV than the majority of currently known bat SARSr-CoVs discovered from other locations, except YNLF_31C and 34C which were recently reported in greater horseshoe bats from another location in Yunnan [22] (Fig 6). The phylogeny of SARSr-CoVs in ORF1a and ORF1b appeared to be associated with their geographical distribution rather than with host species. Regardless of different host bat species, SARS-CoV and SARSr-CoVs detected in bats from southwestern China (Yunnan, Guizhou and Guangxi province) formed one clade, in which SARSr-CoV strains showing closer relationship to SARS-CoV were all from Yunnan. SARSr-CoVs detected in southeastern, central and northern provinces, such as Hong Kong, Hubei and Shaanxi, formed the other clade which was phylogenetically distant to human and civet SARS-CoVs (Fig 6 and S6 Fig).
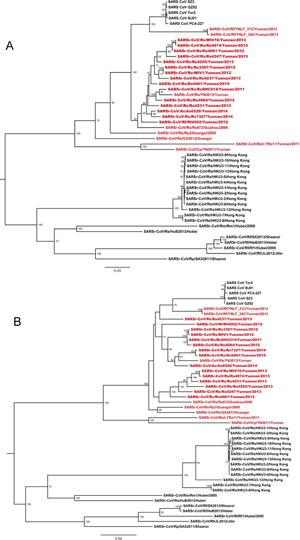
Fig 6. Phylogenetic trees based on nucleotide sequences of ORF1a (A) and ORF1b (B). The trees were constructed by the maximum likelihood method using the LG model with bootstrap values determined by 1000 replicates. Only bootstraps > 50% are shown. The scale bars represent 0.03 (A) and 0.02 (B) substitutions per nucleotide position. Rs, Rhinolophus sinicus; Rf, Rhinolophus ferremequinum; Rm, Rhinolophus macrotis; Ra, Rhinolophus affinis; Rp, Rhinolophus pusillus; As, Aselliscus stoliczkanus; Cp, Chaerephon plicata. SARSr-CoVs detected in bats from the single cave surveyed in this study are in bold. Sequences detected in southwestern China are indicated in red.
Rescue of bat SARSr-CoVs and virus infectivity experiments
In the current study, we successfully cultured an additional novel SARSr-CoV Rs4874 from a single fecal sample using an optimized protocol and Vero E6 cells [17]. Its S protein shared 99.9% aa sequence identity with that of previously isolated WIV16 and it was identical to WIV16 in RBD. Using the reverse genetics technique we previously developed for WIV1 [23], we constructed a group of infectious bacterial artificial chromosome (BAC) clones with the backbone of WIV1 and variants of S genes from 8 different bat SARSr-CoVs. Only the infectious clones for Rs4231 and Rs7327 led to cytopathic effects in Vero E6 cells after transfection (S7 Fig). The other six strains with deletions in the RBD region, Rf4075, Rs4081, Rs4085, Rs4235, As6526 and Rp3 (S1 Fig) failed to be rescued, as no cytopathic effects was observed and viral replication cannot be detected by immunofluorescence assay in Vero E6 cells (S7 Fig). In contrast, when Vero E6 cells were respectively infected with the two successfully rescued chimeric SARSr-CoVs, WIV1-Rs4231S and WIV1-Rs7327S, and the newly isolated Rs4874, efficient virus replication was detected in all infections (Fig 7). To assess whether the three novel SARSr-CoVs can use human ACE2 as a cellular entry receptor, we conducted virus infectivity studies using HeLa cells with or without the expression of human ACE2. All viruses replicated efficiently in the human ACE2-expressing cells. The results were further confirmed by quantification of viral RNA using real-time RT-PCR (Fig 8).
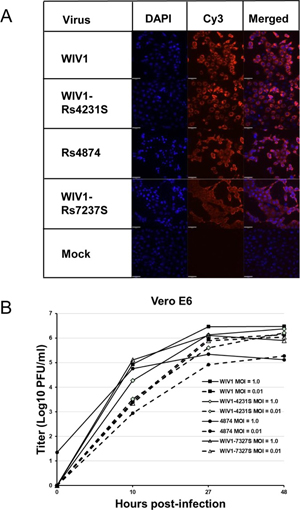
Fig 7. Infection of Vero E6 cells by bat SARSr-CoV WIV1, Rs4874, WIV1-Rs4231S and WIV1-Rs7327S.
(A) The successful infection was confirmed by immunofluorescent antibody staining using rabbit antibody against the SARSr-CoV Rp3 nucleocapsid protein. The columns (from left to right) show staining of nuclei (blue), virus replication (red), and both nuclei and virus replication (merged double-stain images). (B) The growth curves in Vero E6 cells with a MOI of 1.0 and 0.01.
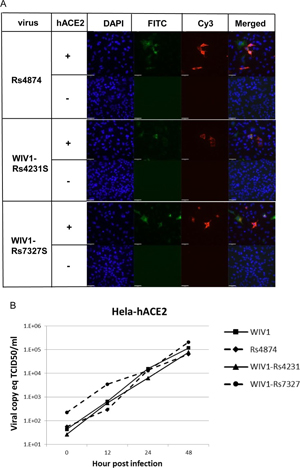
Fig 8. Analysis of receptor usage by immunofluorescence assay (A) and real-time PCR (B). Virus infectivity of Rs4874, WIV1-Rs4231S and WIV1-Rs7327S was determined in HeLa cells with and without the expression of human ACE2. ACE2 expression was detected with goat anti-human ACE2 antibody followed by fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat IgG. Virus replication was detected with rabbit antibody against the SARSr-CoV Rp3 nucleocapsid protein followed by cyanine 3 (Cy3)-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI (49,6-diamidino-2-phenylindole).The columns (from left to right) show staining of nuclei (blue), ACE2 expression (green), virus replication (red) and the merged triple-stained images, respectively.
Activation of activating transcription factor 6 (ATF6) by the ORF8 proteins of different bat SARSr-CoVs
The induction of the ATF6-dependent transcription by the ORF8s of SARS-CoV and bat SARSr-CoVs were investigated using a luciferase reporter, 5×ATF6-GL3. In HeLa cells transiently transfected with the expression plasmids of the ORF8s of bat SARSr-CoV Rf1, Rf4092 and WIV1, the relative luciferase activities of the 5×ATF6-GL3 reporter was enhanced by 5.56 to 9.26 folds compared with cells transfected with the pCAGGS empty vector, while it was increased by 4.42 fold by the SARS-CoV GZ02 ORF8. As a control, the treatment with tunicamyxin (TM) stimulated the transcription by about 11 folds (Fig 9A). The results suggests that various ORF8 proteins of bat SARSr-CoVs can activate ATF6, and those of some strains have a stronger effect than the SARS-CoV ORF8.
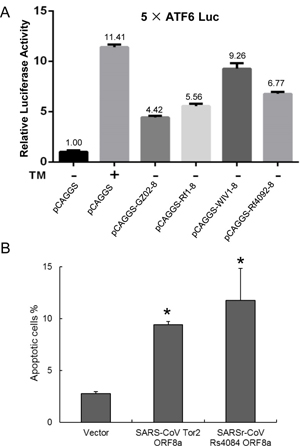
Fig 9. Functional characterization of diverse ORF8 and ORF8a proteins of bat SARSr-CoVs.
(A) The ORF8 proteins of SARS-CoV and bat SARSr-CoVs induces the ATF6-dependent transcriptional activity. HeLa cells were transiently transfected with the pcAGGS expression plasmids of the ORF8 of SARS-CoV GZ02, bat SARSr-CoV Rf1, WIV1 and Rf4092 and the reporter plasmid 5×ATF6-GL3 for 40h. Control cells were co-transfected with the reporter plasmid and the empty pCAGGS vector for 24h, and treated with or without TM (2μg/ml) for an additional 16h. The cell lysates were harvested for dual luciferase assay and data are shown as the average values from triplicate wells. (B) The ORF8a proteins of SARS-CoV and bat SARSr-CoV triggered apoptosis. 293T cells were transfected with the expression plasmids of the ORF8a of SARS-CoV Tor2 and bat SARSr-CoV Rs4084 and a pcAGGS vector control for 24h. Apoptosis was analyzed by flow cytometry after annexin V staining and the percentage of apoptotic cells were calculated. Data are shown as the average values from triplicate cells. Error bars indicate SDs. * P<0.05.
Induction of apoptosis by the ORF8a of the newly identified bat SARSr-CoV
We conducted transient transfection to examine whether the ORF8a of SARSr-CoV Rs4084 triggered apoptosis. As shown in Fig 9B, 11.76% and 9.40% of the 293T cells transfected with the SARSr-CoV Rs4084-ORF8a and SARS-CoV Tor2-ORF8a expression plasmid underwent apoptosis, respectively. In contrast, transfection with the empty vector resulted in apoptosis in only 2.79% of the cells. The results indicate that Rs4084 ORF8a has an apoptosis induction activity similar to that of SARS-CoV [28].
Discussion
Genetically diverse SARSr-CoVs have been detected in various horseshoe bat species across a wide geographic range in China in the past decade [9–12,14,29]. However, most bat SARSr-CoVs show considerable genetic distance to SARS-CoV, particularly in the highly variable S1, ORF8 and ORF3 regions [10,25]. Recently, several novel SARSr-CoVs have been described to be more closely related to SARS-CoV, either in the S gene or in ORF8. The S proteins of RsSHC014, Rs3367, WIV1 and WIV16, which were reported in our previous studies, shared 90% to 97% aa sequence identities to those of human/civet SARS-CoVs [17,18]. Another strain from Rhinolophus affinis in Yunnan termed LYRa11 showed 90% aa sequence identity to SARS-CoV in the S gene [13]. In addition, two studies have described 4 novel SARSr-CoVs (YNLF_31C/34C and GX2013/YN2013) which possessed a full-length ORF8 with substantially higher similarity to that of SARS-CoV [22,30]. These findings provide strong genetic evidence for the bat origin of SARS-CoV with regard to the S gene or ORF8. However, all of these SARSr-CoVs were distinct from SARS-CoV in at least one other gene, suggesting that none of them was the immediate progenitor of SARS-CoV. Moreover, these SARSr-CoVs were discovered in bat populations from physically distinct locations. The site of origin of the true progenitor of SARS-CoV and the evolutionary origin of SARS-CoV have until now remained elusive. In the current study, we have identified a bat habitat potentially important for SARSr-CoV evolution where a series of recombination events have likely occurred among different SARSr-CoV strains, which provides new insights into the origin of SARS-CoV.
SARS first emerged in Guangdong province in late 2002 [7]. However, SARSr-CoVs discovered in bats from neighboring areas of Guangdong to date have shown phylogenetic disparity from SARS-CoV especially in the S gene [9,10,14], suggesting SARS-CoV may have originated from another region. Our analysis of the phylogeny of SARS-CoVs and all known bat SARSr-CoVs using the nt sequence of their non-structural ORF1a and ORF1b genes, which constitute the majority of the genome, shows that SARSr-CoV evolution is strongly correlated with their geographical origin, but not host species. It is noteworthy that SARSr-CoVs detected in Yunnan are more closely related to SARS-CoV than strains from other regions in China. This finding implies that Yunnan, or southwestern China, is more likely to be the geographical source of SARS-CoV than other regions in China, but data from more extensive surveillance are yet needed to support this inference.
In our longitudinal surveillance of SARSr-CoVs in a single cave in Yunnan where we discovered Rs3367, RsSHC014, WIV1 and WIV16, the CoV prevalence in fecal samples varied among different sampling time. Generally, a higher prevalence was observed in autumn (September and October) than in spring and early summer (April and May). This may be due to the establishment of a susceptible subpopulation of newborn bats which had not developed their own immunity after the parturition period [31]. Another factor may be the changes in the composition of bat species in the cave at different sampling dates. For example, in September 2012 when the CoV prevalence reached 51.3%, the majority of samples were from R. sinicus, but in May 2015 when only 3 out of the 145 samples tested positive, Aselliscus stoliczkanus was the predominant bat species in the cave. We failed to amplify the RBD sequences from 15 of the 64 SARSr-CoV positive samples. Most of these samples had comparatively low viral concentration (< 107 copies/g) (S8 Fig), as revealed by our previous quantitative studies [32]. The unsuccessful amplification of RBD in some samples with high viral concentration was probably because of the more divergent sequences in this region of these SARSr-CoV genomes.
In this cave, we have now obtained full-length genome sequences of additional 11 novel SARSr-CoVs from bats. Our findings suggest the co-circulation of different bat SARSr-CoVs highly similar to SARS-CoV in the most variable S1 (NTD and RBD), ORF8 and ORF3 regions, respectively, in this single location. In the ORF1a, ORF1b, E, M and N genes, the SARSr-CoVs circulating in this cave also shared > 98% aa sequence identities with human/civet SARS-CoVs. Thus, all of the building blocks of the SARS-CoV genome were present in SARSr-CoVs from this single location in Yunnan during our sampling period. Furthermore, strains closely related to different representative bat SARSr-CoVs from other provinces (e.g. Rs672, HKU3 and Rf1) in the RBD region were also detected there. Therefore, this cave could be regarded as a rich gene pool of bat SARSr-CoVs, wherein concurrent circulation of a high diversity of SARSr-CoV strains has led to an unusually diverse assemblage of SARSr-CoVs.
During our 5-year surveillance in this single cave, we first reported Rs3367 and WIV1 in 2013, with RBD sequence closely resembling that of SARS-CoV [17]. More recently, we discovered WIV16 which had an RBD almost identical to WIV1’s but shared much higher similarity with SARS-CoV than WIV1 in the NTD region of S1, making it the closest SARSr-CoV to the epidemic strains identified to date [18]. In this study, we found a novel strain Rs4231 from the same location sharing almost identical NTD sequence with WIV16 but distinct from it in the RBD, with evidence of a recombination event. Our recombination analysis indicated that a recombination event may have taken place at the junction between the coding region of NTD and RBD in the Rs4231 and WIV1 genomes and resulted in WIV16. Recombination at this genomic position also happened among other SARSr-CoVs relatively distant to SARS-CoV found in this location (e.g. Rs4081 and Rs4247, S5 Fig). The frequent recombination at this hotspot in the S gene increased the genetic diversity of SARSr-CoVs harbored in these bat populations and might have been responsible for the generation of the S gene of the direct progenitor strain of SARS-CoV.
The genomes of SARS-CoVs from patients during the early epidemic phase and civet SARS-CoVs all contained a single full-length ORF8 [3,7]. We have found that a number of bat SARSr-CoVs from this cave possessed a complete ORF8 highly similar to that of early human/civet SARS-CoV (>97% nt sequence identity), represented by strain Rf4092 (S3C Fig). This provided further evidence for the source of human SARS-CoV ORF8 in bats [22,30]. In contrast, the ORF8 was split into overlapping ORF8a and ORF8b in most human SARS-CoV strains from later-phase patients due to the acquisition of a 29-nt deletion [8,26]. In this study, we have discovered for the first time a bat SARSr-CoV with ORF8a and ORF8b highly similar to the later-phase human SARS-CoVs, though the split of ORF8 in the bat SARSr-CoV and that in human SARS-CoV were two independent events. Our recombination analysis suggests that this strain, Rs4084, likely acquired its ORF8 from Rf4092 through recombination, followed by the development of the 5-nt deletion which led to the splitting. It suggests that ORF8 region in bat SARSr-CoV genomes is prone to deletions as in human SARS-CoV [3,25]. Finally, the recombination analysis suggests that an ancestral strain of SARS-CoV SZ3 would have been generated if the recombination around ORF8 had occurred between the lineages that led to WIV16 and Rf4092. Taken together, the evidence of recombination events among SARSr-CoVs harbored by bats in this single location suggests that the direct progenitor of SARS-CoV may have originated as a result of a series of recombination within the S gene and around ORF8. This could have been followed by the spillover from bats to civets and people either in the region, or during movement of infected animals through the wildlife trade. However, given the paucity of data on animal trade prior to the SARS outbreak, the likely high geographical sampling bias in bat surveillance for SARSr-CoVs in southern China, and the possibility that other caves harbor similar bat species assemblages and a rich diversity of SARSr-CoVs, a definite conclusion about the geographical origin of SARS-CoV cannot be drawn at this point.
R. sinicus are regarded as the primary natural host of SARS-CoV, as all SARSr-CoVs highly homologous to SARS-CoV in the S gene were predominantly found in this species. However, it is noted that two SARSr-CoVs previously reported from R. ferrumequinum showed the closest phylogenetic position to SARS-CoV in the ORF1a/1b trees. These strains were discovered in another location in Yunnan 80 km from the cave surveyed in the current study [22]. This information also supports the speculation that SARS-CoV may have originated from this region. Nonetheless, since the correlation between the host species and the phylogeny of SARSr-CoV ORF1ab seems limited, more SARSr-CoV sequences need to be obtained from different Rhinolophus bat species in both locations in Yunnan, and from other locations in southern China. In particular, it will be important to assess whether R. ferrumequinum played a more important role in the evolution of SARS-CoV ORF1ab.
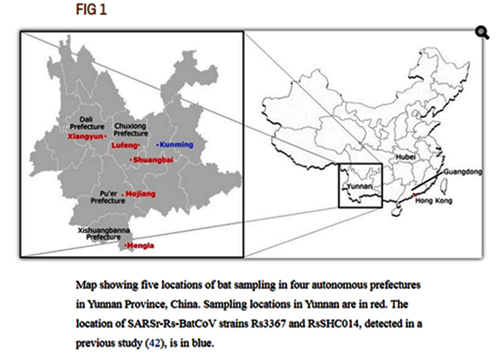
The cave we studied is located approximately 60 km from the city of Kunming. Beside a number of rhinolophid and hipposiderid species from which SARSr-CoVs have been detected, other bats like myotis were also present there. The temperature in the cave is around 22–25°C and the humidity around 85%-90%. The physical nature of the cave is not unique, but it does appear to host a particularly dense population of bats in the reproductive season. Similar caves co-inhabited by bat populations of different species are not rare in other areas in Yunnan. We propose that efforts to study the ecology, host species diversity, and viral strain populations of these caves may provide critical information on what drives SARSr-CoV evolution.
Our previous studies demonstrated the capacity of both WIV1 and WIV16 to use ACE2 orthologs for cell entry and to efficiently replicate in human cells [17,18]. In this study, we confirmed the use of human ACE2 as receptor of two novel SARSr-CoVs by using chimeric viruses with the WIV1 backbone replaced with the S gene of the newly identified SARSr-CoVs. Rs7327’s S protein varied from that of WIV1 and WIV16 at three aa residues in the receptor-binding motif, including one contact residue (aa 484) with human ACE2. This difference did not seem to affect its entry and replication efficiency in human ACE2-expressing cells. A previous study using the SARS-CoV infectious clone showed that the RsSHC014 S protein could efficiently utilize human ACE2 [33], despite being distinct from SARS-CoV and WIV1 in the RBD (S1 Fig). We examined the infectivity of Rs4231, which shared similar RBD sequence with RsSHC014 but had a distinct NTD sequence, and found the chimeric virus WIV1-Rs4231S also readily replicated in HeLa cells expressing human ACE2 molecule. The novel live SARSr-CoV we isolated in the current study (Rs4874) has an S gene almost identical to that of WIV16. As expected, it is also capable of utilizing human ACE2. These results indicate that diverse variants of SARSr-CoV S protein without deletions in their RBD are able to use human ACE2. In contrast, our previous study revealed that the S protein of a R. sinicus SARSr-CoV with deletions (Rp3) failed to use human, civet and bat ACE2 for cell entry [34]. In this study, in addition to Rs4231 and Rs7327, we also constructed infectious clones with the S gene of Rs4081, Rf4075, Rs4085, Rs4235 and As6526, which all contained the deletions in their RBD. These 7 strains, plus Rs4874 and the previously studied WIV1 and RsSHC014, could represent all types of S variants of SARSr-CoVs in this location (S3A Fig). However, none of the strains with deletions in the RBD could be rescued from Vero E6 cells. Therefore, the two distinct clades of SARSr-CoV S gene may represent the usage of different receptors in their bat hosts.
The full-length ORF8 protein of SARS-CoV is a luminal endoplasmic reticulum (ER) membrane-associated protein that induces the activation of ATF6, an ER stress-regulated transcription factor that activates the transcription of ER chaperones involved in protein folding [35]. We amplified the ORF8 genes of Rf1, Rf4092 and WIV1, which represent three different genotypes of bat SARSr-CoV ORF8 (S3C Fig), and constructed the expression plasmids. All of the three ORF8 proteins transiently expressed in HeLa cells can stimulate the ATF6-dependent transcription. Among them, the WIV1 ORF8, which is highly divergent from the SARS-CoV ORF8, exhibited the strongest activation. The results indicate that the variants of bat SARSr-CoV ORF8 proteins may play a role in modulating ER stress by activating the ATF6 pathway. In addition, the ORF8a protein of SARS-CoV from the later phase has been demonstrated to induce apoptosis [28]. In this study, we have found that the ORF8a protein of the newly identified SARSr-CoV Rs4084, which contained an 8-aa insertion compared with the SARS-CoV ORF8a, significantly triggered apoptosis in 293T cells as well.
Compared with the 154-aa ORF3b of SARS-CoV, the ORF3b proteins of all previously identified bat SARSr-CoVs were smaller in size due to the early translation termination. However, for the first time, we discovered an ORF3b without the C-terminal truncation in a bat SARSr-CoV, Rs7327, which differed from the ORF 3b of SARS-CoV GZ02 strain at only one aa residue. The SARS-CoV ORF3b antagonizes interferon function by modulating the activity of IFN regulatory factor 3 (IRF3) [27]. As previous studies suggested, the nuclear localization signal-containing C-terminal may not be required for the IFN antagonist activity of ORF3b [36]. Our previous studies also demonstrated that the ORF3b protein of a bat SARSr-CoV, termed Rm1, which was C-terminally truncated to 56 aa and shared 62% aa sequence identity with SARS-CoV, still displayed the IFN antagonist activity [37]. It is very interesting to investigate in further studies whether Rs7327’s ORF3b and other versions of truncated ORF3b such as WIV1 and WIV16 also show IFN antagonism profiles.
As a whole, our findings from a 5-year longitudinal study conclusively demonstrate that all building blocks of the pandemic SARS-CoV genome are present in bat SARSr-CoVs from a single location in Yunnan. The data show that frequent recombination events have happened among those SARSr-CoVs in the same cave. While we cannot rule out the possibility that similar gene pools of SARSr-CoVs exist elsewhere, we have provided sufficient evidence to conclude that SARS-CoV most likely originated from horseshoe bats via recombination events among existing SARSr-CoVs. In addition, we have also revealed that various SARSr-CoVs capable of using human ACE2 are still circulating among bats in this region. Thus, the risk of spillover into people and emergence of a disease similar to SARS is possible. This is particularly important given that the nearest village to the bat cave we surveyed is only 1.1 km away, which indicates a potential risk of exposure to bats for the local residents. Thus, we propose that monitoring of SARSr-CoV evolution at this and other sites should continue, as well as examination of human behavioral risk for infection and serological surveys of people, to determine if spillover is already occurring at these sites and to design intervention strategies to avoid future disease emergence.
Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus
by Ben Hu, Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – original draft,#1 Lei-Ping Zeng, Investigation, Methodology, Xing-Lou Yang, Investigation, Resources, Xing-Yi Ge, Formal analysis, Resources, Wei Zhang, Investigation, Bei Li, Investigation, Jia-Zheng Xie, Investigation, Xu-Rui Shen, Investigation, Yun-Zhi Zhang, Resources, Ning Wang, Investigation,1 Dong-Sheng Luo, Investigation, Resources,1 Xiao-Shuang Zheng, Investigation, Mei-Niang Wang, Resources,1 Peter Daszak, Funding acquisition, Writing – review & editing, Lin-Fa Wang, Conceptualization, Funding acquisition, Writing – review & editing, Jie Cui, Conceptualization, Formal analysis, Funding acquisition, Software, Writing – review & editing, and Zheng-Li Shi, Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – review & editing
Christian Drosten, Editor
Copyright © 2017 Hu et al
NOTICE: THIS WORK MAY BE PROTECTED BY COPYRIGHT
YOU ARE REQUIRED TO READ THE COPYRIGHT NOTICE AT THIS LINK BEFORE YOU READ THE FOLLOWING WORK, THAT IS AVAILABLE SOLELY FOR PRIVATE STUDY, SCHOLARSHIP OR RESEARCH PURSUANT TO 17 U.S.C. SECTION 107 AND 108. IN THE EVENT THAT THE LIBRARY DETERMINES THAT UNLAWFUL COPYING OF THIS WORK HAS OCCURRED, THE LIBRARY HAS THE RIGHT TO BLOCK THE I.P. ADDRESS AT WHICH THE UNLAWFUL COPYING APPEARED TO HAVE OCCURRED. THANK YOU FOR RESPECTING THE RIGHTS OF COPYRIGHT OWNERS.
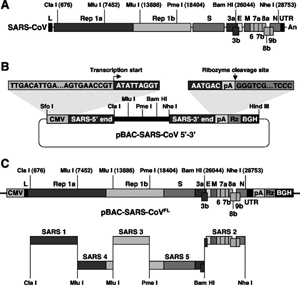
There have been three publications, in 2015,30 2016 and 2017, describing the WIV gain of function research. The WIV, having learned both basic and traceless infectious-clone technology from joint research with a laboratory at the University of North Carolina (UNC) in 2015, initiated construction of novel chimeric coronaviruses without UNC immediately thereafter. WIV’s first publication on the use of basic infectious-clone technology to construct novel chimeric coronaviruses at WIV appeared in 2016.31 WIV’s first publication on the use of traceless, signature-free infectious-clone technology also appeared in 2016.32
-- Did the SARS-CoV-2 virus arise from a bat coronavirus research program in a Chinese laboratory? Very possibly, by Milton Leitenberg
Abstract
A large number of SARS-related coronaviruses (SARSr-CoV) have been detected in horseshoe bats since 2005 in different areas of China. However, these bat SARSr-CoVs show sequence differences from SARS coronavirus (SARS-CoV) in different genes (S, ORF8, ORF3, etc) and are considered unlikely to represent the direct progenitor of SARS-CoV. Herein, we report the findings of our 5-year surveillance of SARSr-CoVs in a cave inhabited by multiple species of horseshoe bats in Yunnan Province, China. The full-length genomes of 11 newly discovered SARSr-CoV strains, together with our previous findings, reveals that the SARSr-CoVs circulating in this single location are highly diverse in the S gene, ORF3 and ORF8. Importantly, strains with high genetic similarity to SARS-CoV in the hypervariable N-terminal domain (NTD) and receptor-binding domain (RBD) of the S1 gene, the ORF3 and ORF8 region, respectively, were all discovered in this cave. In addition, we report the first discovery of bat SARSr-CoVs highly similar to human SARS-CoV in ORF3b and in the split ORF8a and 8b. Moreover, SARSr-CoV strains from this cave were more closely related to SARS-CoV in the non-structural protein genes ORF1a and 1b compared with those detected elsewhere. Recombination analysis shows evidence of frequent recombination events within the S gene and around the ORF8 between these SARSr-CoVs. We hypothesize that the direct progenitor of SARS-CoV may have originated after sequential recombination events between the precursors of these SARSr-CoVs. Cell entry studies demonstrated that three newly identified SARSr-CoVs with different S protein sequences are all able to use human ACE2 as the receptor, further exhibiting the close relationship between strains in this cave and SARS-CoV. This work provides new insights into the origin and evolution of SARS-CoV and highlights the necessity of preparedness for future emergence of SARS-like diseases.
Author summary
Increasing evidence has been gathered to support the bat origin of SARS coronavirus (SARS-CoV) in the past decade. However, none of the currently known bat SARSr-CoVs is thought to be the direct ancestor of SARS-CoV. Herein, we report the identification of a diverse group of bat SARSr-CoVs in a single cave in Yunnan, China. Importantly, all of the building blocks of SARS-CoV genome, including the highly variable S gene, ORF8 and ORF3, could be found in the genomes of different SARSr-CoV strains from this single location. Based on the analysis of full-length genome sequences of the newly identified bat SARSr-CoVs, we speculate that the direct ancestor of SARS-CoV may have arisen from sequential recombination events between the precursors of these bat SARSr-CoVs prior to spillover to an intermediate host. In addition, we found bat SARSr-CoV strains with different S proteins that can all use the receptor of SARS-CoV in humans (ACE2) for cell entry, suggesting diverse SARSr-CoVs capable of direct transmission to humans are circulating in bats in this cave. Our current study therefore offers a clearer picture on the evolutionary origin of SARS-CoV and highlights the risk of future emergence of SARS-like diseases.
Introduction
Severe Acute Respiratory Syndrome (SARS) is a severe emerging viral disease with high fatality characterized by fever, headache and severe respiratory symptoms including cough, dyspnea and pneumonia [1]. Due to its high transmissibility among humans, after its first emergence in southern China in late 2002, it rapidly led to a global pandemic in 2003 and was marked as one of the most significant public health threats in the 21st century [2,3]. The causative agent, SARS coronavirus (SARS-CoV), has been previously assigned to group 2b CoV and is now a member of the lineage B of genus Betacoronavirus in the family Coronaviridae [4]. It shares similar genome organization with other coronaviruses, but exhibits a unique genomic structure which includes a number of specific accessory genes, including ORF3a, 3b, ORF6, ORF7a, 7b, ORF8a, 8b and 9b [5,6].
Masked palm civets (Paguma larvata) were initially hypothesized to be the animal origin of SARS-CoV [7,8]. However, since a large number of genetically diverse SARS-related coronaviruses (SARSr-CoV) have been detected in multiple species of horseshoe bats (genus Rhinolophus) from different areas of China and Europe in the aftermath of SARS, it is prevailingly considered that SARS-CoV originated in horseshoe bats with civets acting as the intermediate amplifying and transmitting host [9–16]. Recently we have reported four novel SARSr-CoVs from Chinese horseshoe bats that shared much higher genomic sequence similarity to the epidemic strains, particularly in their S gene, of which two strains (termed WIV1 and WIV16) have been successfully cultured in vitro [17,18]. These newly identified SARSr-CoVs have been demonstrated to use the same cellular receptor (angiotensin converting enzyme-2 [ACE-2]) as SARS-CoV does and replicate efficiently in primary human airway cells [17–19].
Despite the cumulative evidence for the emergence of SARS-CoV from bats, all bat SARSr-CoVs described so far are clearly distinct from SARS-CoV in the S gene and/or one or more accessory genes such as ORF3 and ORF8, suggesting they are likely not the direct ancestor of SARS-CoV. Thus a critical gap remains in our understanding of how and where SARS-CoV originated from bat reservoirs. Previously, we reported a number of bat SARSr-CoVs with diverse S protein sequences from a single cave in Yunnan Province, including the four strains mentioned above most closely related to SARS-CoV [17,18]. Here we report the latest results of our 5-year longitudinal surveillance of bat SARSr-CoVs in this single location and systematic evolutionary analysis using full-length genome sequences of 15 SARSr-CoV strains (11 novel ones and 4 from previous studies). Efficiency of human ACE2 usage and the functions of accessory genes ORF8 and 8a were also evaluated for some of the newly identified strains.
Results
Continued circulation of diverse SARSr-CoVs in bats from a single location
We have carried out a five-year longitudinal surveillance (April 2011 to October 2015) on SARSr-CoVs in bats from a single habitat in proximity to Kunming city, Yunnan province, China, which was mainly inhabited by horseshoe bats. A total of 602 alimentary specimens (anal swabs or feces) were collected and tested for the presence of CoVs by a Pan-CoV RT-PCR targeting the 440-nt RdRp fragment that is conserved among all known α- and β-CoVs [20]. In total, 84 samples tested positive for CoVs. Sequencing of the PCR amplicons revealed the presence of SARSr-CoVs in the majority (64/84) of the CoV-positive samples (Table 1). Host species identification by amplification of either Cytb or ND1 gene suggested that most (57/64) of the SARSr-CoV positive samples were from Rhinolophus sinicus, while the remaining 7 samples were from Rhinolophus ferrumequinum, Rhinolophus affinis and from Aselliscus stoliczkanus which belongs to the family Hipposideridae.
Table 1: Summary of SARSr-CoV detection in bats from a single habitat in Kunming, Yunnan.
Sampling time / Sample type / Sample Numbers / SARSr-CoV + bat species (No.)
Total CoV + SARSr-CoV +
April, 2011 / anal swab / 14 / 1 / 1 / R. sinicus (1)
October, 2011 / anal swab / 8 / 3 / 3 / R. sinicus (3)
May, 2012 / anal swab & feces / 54 / 9 / 4 / R. sinicus (4)
September, 2012 / feces / 39 / 20 / 19 / R. sinicus (16); R. ferrumequinum (3)
April, 2013 / feces / 52 / 21 / 16 / R. sinicus (16)
July, 2013 / anal swab & feces / 115 / 9 / 8 / R. sinicus (8)
May, 2014 / feces / 131 / 8 / 4 / A. stoliczkamus (3); R. affinis (1)
October, 2014 / anal swab / 19 / 4 / 4 / R. sinicus (4)
May, 2015 / feces / 145 / 3 / 0 / --
October, 2015 / anal swab / 25 / 6 / 5 / R. sinicus (5)
Total / -- / 602 / 84 / 64 / R (61) A (3)
Based on the preliminary analysis of the partial RdRp sequences, all of the 64 bat SARSr-CoV sequences showed high similarity among themselves and with other reported bat SARSr-CoVs and SARS-CoVs from humans and civets. To understand the genetic diversity of these bat SARSr-CoVs, the most variable region of the SARSr-CoV S gene, corresponding to the receptor-binding domain (RBD) of SARS-CoV, were amplified and sequenced. Due to low viral load in some samples, RBD sequences were successfully amplified only from 49 samples. These RBD sequences displayed high genetic diversity and could be divided into two large clades, both of which included multiple genotypes. Clade 1 strains shared an identical size and higher amino acid (aa) sequence identity with SARS-CoV RBD, while clade 2 had a shorter size than SARS-CoV S due to two deletions (5 and 12–13 aa, respectively) (S1 Fig). Co-infections by two strains of different clades were detected in two samples, Rs3262 and Rs4087 (S1 Fig).
Genomic characterization of the novel SARSr-CoVs
Based on the diversity of RBD sequences, 11 novel SARSr-CoV strains named by abbreviation of bat species and sample ID (Rs4081, Rs4084, Rs4231, Rs4237, Rs4247, Rs4255, Rs4874, Rs7327, Rs9401, Rf4092 and As6526) were selected for full-length genomic sequencing based on sample abundance, genotype of RBD as well as sampling time. For each RBD genotype and each time of sampling, at least one representative strain was selected. The genome size of these novel SARSr-CoVs ranged from 29694 to 30291 nucleotides (nt). This gave a total of 15 full-length genomes of bat SARSr-CoVs from this single location (13 from R.sinicus, and one each from R. ferrumequinum and A. stoliczkanus), including our previously reported strains, Rs3367, RsSHC014, WIV1 and WIV16 [17,18]. The genomes of all 15 SARSr-CoVs circulating in this single cave shared 92.0% to 99.9% nt sequence identity. The overall nt sequence identity between these SARSr-CoVs and human and civet SARS-CoVs is 93.2% to 96%, significantly higher than that observed for bat SARSr-CoVs reported from other locations in China (88–93%) [9,10,12,14,21,22]. The genome sequence similarity among the 15 SARSr-CoVs and SARS-CoV SZ3 strain was examined by Simplot analysis (Fig 1). The 15 SARSr-CoVs are highly conserved and share a uniformly high sequence similarity to SARS-CoV in the non-structural gene ORF1a (96.6% to 97.1% nt sequence identity, 98.0% to 98.3% aa sequence identity) and ORF1b (96.1% to 96.6% nt sequence identity, 99.0% to 99.4% aa sequence identity). In contrast, a considerable genetic diversity is shown in the S gene (corresponding to SZ3 genome position 21477 to 25244) and ORF8 (corresponding to SZ3 genome position 27764 to 28132) (Fig 1).
Fig 1. Similarity plot based on the full-length genome sequence of civet SARS CoV SZ3.
Full-length genome sequences of all SARSr-CoV detected in bats from the cave investigated in this study were used as reference sequences. The analysis was performed with the Kimura model, a window size of 1500 base pairs and a step size of 150 base pairs.
The 11 novel SARSr-CoVs identified from this single location generally shared similar genome organization with SARS-CoV and other bat SARSr-CoVs. In our previous study, we identified an additional ORF termed ORFx present between ORF6 and ORF7 in strain WIV1 and WIV16 [18,23]. In this study, ORFx was also found in the genomes of Rs7327 and Rs4874. Compared with that of WIV1 and WIV16, the length of ORFx in Rs7327 and Rs4874 was extended to 510 nt due to a deletion of 2 nt in a poly-T sequence that resulted in a shift of reading frame (Fig 2 and S2 Fig).
Fig 2. Schematic diagram illustrating the genomic regions or ORFs with most variation between different SARS-CoV and SARSr-CoV isolates.
Coding regions of the N-terminal domain (NTD) and receptor-binding domain (RBD) of the spike protein, ORF3a/b and ORF8 (8a/b) in bat SARSr-CoV genomes highly similar to those in SARS CoV genome are indicated with black boxes or arrows while the hollow boxes or arrows represent corresponding regions with less sequence similarity to those of SARS-CoV. The deletions in the RBD of some SARSr-CoVs are indicated by two vertical lines.
Co-circulation of different bat SARSr-CoVs with S, ORF8 and ORF3 sequences similar to those in SARS-CoV at a single location
The primary difference between SARS-CoV and most bat SARSr-CoVs is located in S gene. The S protein is functionally divided into two subunits, denoted S1 and S2, which is responsible for receptor binding and cellular membrane fusion, respectively. S1 consists of two domains, the N-terminal domain (NTD) and C-terminal domain (CTD) which is also known as the RBD in SARS-CoV [24]. SARS-CoV and bat SARSr-CoVs share high sequence identity in the S2 region in contrast to the S1 region. Among the 15 SARSr-CoVs identified from bats in the surveyed cave, six strains with deletions in their RBD regions (Rs4081, Rs4237, Rs4247, Rs4255, Rf4092 and As6526) showed 78.2% to 80.2% aa sequence identity to SARS-CoV in the S protein, while the other nine strains without deletions were much more closely related to SARS-CoV, with 90.0% (Rs4084) to 97.2% (Rs4874) aa sequence identity. These nine SARSr-CoVs can be further divided into four genotypes according to their S1 sequences (Fig 2): RsSHC014/Rs4084 showed more genetic differences from SARS-CoV in both NTD and RBD regions; The RBD sequences of SARSr-CoV Rs7327, Rs9401 and previously reported WIV1/Rs3367 closely resembled that of SARS-CoV. However, they were distinct from SARS-CoV but similar to RsSHC014 in NTD. In contrast, we found a novel SARSr-CoV, termed Rs4231, which shared highly similar NTD, but not RBD sequence with SARS-CoV (Figs (Figs22 and and3).3). Its S protein showed 94.6% to 95% aa sequence identity to those of human and civet SARS-CoVs (S1 Table). Strains with both NTD and RBD highly homologous to those of SARS-CoV were also present in this cave. In addition to WIV16 which we described previously [18], Rs4874 was also found to have the S protein closest to SARS-CoV S (> 97% aa sequence identity) of all the bat SARSr-CoVs reported to date (Figs (Figs22 and and3).3). In addition to the SARSr-CoVs subjected to full-length genome sequencing, we also obtained the RBD and NTD sequences from other samples collected in this cave. The sequences with high identity to SARS-CoV RBD were amplified from 10 more R. sinicus samples. SARSr-CoVs with this genotype of RBD were detected in different seasons throughout the five years. Strains containing the NTD similar to SARS-CoV were only found in 2013 (S2 Table).
Fig 3. Amino acid sequence comparison of the S1 subunit (corresponding to aa 1–660 of the spike protein of SARS-CoV).
The receptor-binding domain (aa 318–510) of SARS-CoV and the homologous region of bat SARSr-CoVs are indicated by the red box. The key aa residues involved in the interaction with human ACE2 are numbered on top of the aligned sequences. SARS-CoV GZ02, BJ01 and Tor2 were isolated from patients in the early, middle and late phase, respectively, of the SARS outbreak in 2003. SARS-CoV SZ3 was identified from civets in 2003. SARSr-CoV Rs 672 and YN2013 were identified from R. sinicus collected in Guizhou and Yunnan Province, respectively. SARSr-CoV Rf1 and JL2012 were identified from R. ferrumequinum collected in Hubei and Jilin Province, respectively. WIV1, WIV16, RsSHC014, Rs4081, Rs4084, Rs4231, Rs4237, Rs4247, Rs7327 and Rs4874 were identified from R.sinicus, and Rf4092 from R. ferrumequinum in the cave surveyed in this study.
ORF8 is another highly variable gene among different SARS-CoV and SARSr-CoV strains [25,26]. We aligned the ORF8 nt sequences of the representative SARSr-CoVs discovered in this surveillance with those of other SARSr-CoVs and SARS-CoVs (Fig 4). Though WIV16, WIV1, Rs4231 and RsSHC014 were genetically closer to SARS-CoV in S gene, they contained a single 366-nt ORF8 without the 29-nt deletion present in most human SARS-CoVs and showed only 47.1% to 51.0% nt sequence identity to human and civet SARS-CoVs. However, the ORF8 of strain Rf4092 from R. ferrumequinum exhibited high similarity to that of civet SARS-CoV. It possessed a single long ORF8 of the same length (369 nt) as that of civet SARS-CoV strain SZ3, with only 10 nt mutations and 3 aa mutations detected (Fig 4). Similar ORF8 sequences were also amplified from other 7 samples collected in the cave during 2011 to 2013, from both R. ferrumequinum and R. sinicus (S2 Table). The ORF8 of Rs4084 was highly similar to Rf4092’s but was split into two overlapping ORFs, ORF8a and ORF8b, due to a short 5-nt deletion (Figs (Figs22 and and4).4). The position of start codons and stop codons of the two ORFs were consistent with those in most human SARS-CoV strains. Excluding the 8-aa insertion, Rs4084 and SARS-CoV strain BJ01 displayed identical aa sequence of ORF8a, and only three different aa residues were observed between their ORF8b (Fig 4). To our knowledge, Rs4084 was the first bat SARSr-CoV reported that resembled the late human SARS-CoVs in both ORF8 gene organization and sequence.
Fig 4. Alignment of nucleotide sequences of ORF8 or ORF8a/8b.
The start codons and stop codons of ORF8, 8a and 8b are marked with black boxes and the forward and reverse arrows, respectively. The deletion responsible for the split ORF8a and 8b in human SARS-CoV BJ01, Tor2 and bat SARSr-CoV Rs4084 is marked with red boxes. See the legend for Fig 3 for the origin of various sequences used in this alignment.
Another key difference between SARS-CoV and bat SARSr-CoV genomes is the ORF3 coding region [10,17,21]. We analyzed the ORF3a sequences amplified from 42 samples and found that most of the SARSr-CoVs closely related to SARS-CoV in the S gene shared higher ORF3a sequence similarity (96.4% to 98.9% aa identity) with SARS-CoV (S3 Fig and S2 Table). The ORF3b of SARS CoV, sharing a large part of its coding sequence with the ORF3a, encodes a 154-aa protein [27], but it is truncated to different extents at the C-terminal in previously described bat SARSr-CoVs including WIV1 and WIV16 (S4 Fig). In the current study, we identified a non-truncated ORF3b for the first time (Rs7327), which maintained the nuclear localization signal at its C-terminal. Moreover, it shared 98.1% aa sequence identity with SARS-CoV strain Tor2 with only three aa substitutions (S4 Fig). Thus, Rs7327 is the bat SARSr-CoV most similar to SARS-CoV in the ORF3 region known to date.
Recombination analysis
The full-length genome sequences of all 15 SARSr-CoVs from the surveyed cave were screened for evidence of potential recombination events. Both similarity plot and bootscan analyses revealed frequent recombination events among these SARSr-CoV strains. It was suggested that WIV16, the closest progenitor of human SARS-CoV known to date [18], was likely to be a recombinant strain from three SARSr-CoVs harbored by bats in the same cave, namely WIV1, Rs4231 and Rs4081, with strong P value (<10−30). Breakpoints were identified at genome positions nt 18391, 22615 and 28160 (Fig 5A). In the genomic region between nt 22615 and 28160, which contained the region encoding the RBD and the S2 subunit of the S protein, WIV16 was highly similar to WIV1, sharing 99% sequence identity. In contrast, in the region between nt 18391 and 22615, which covered a part of ORF1b and the region encoding the NTD of the S gene, WIV16 showed substantially closer relationship to Rs4231. Meanwhile, the ORF1ab sequences upstream from nt 18391 of WIV16 displayed the highest genetic similarity (99.8% nt sequence identity) to that of Rs4081.
Fig 5. Detection of potential recombination events by similarity plot and boot scan analysis.
(A) Full-length genome sequence of SARSr-CoV WIV16 was used as query sequence and WIV1, Rs4231 and Rs4081 as reference sequences. (B) Full-length genome sequence of SARS-CoV SZ3 was used as query sequence and SARSr-CoV WIV16, Rf4092 and Rs4081 as reference sequences. All analyses were performed with a Kimura model, a window size of 1500 base pairs, and a step size of 150 base pairs. The gene map of query genome sequences are used to position breakpoints.
Evidence of recombination event was also detected in the genome of the novel SARSr-CoV Rs4084, which had a unique genome organization with split ORF8a and 8b. The previously reported strain RsSHC014 and the newly identified strain Rf4092 were suggested to be the major and minor parent of Rs4084, respectively (P value < 10−80). The breakpoint was located at nt 26796 (S5 Fig). In the region downstream of the breakpoint including ORF8, Rs4084 showed closet genetic relationship with Rf4092, sharing 98.9% nt sequence identity, while it shared the highest nt sequence identity (99.4%) with RsSHC014 in the majority of its genome upstream from the breakpoint.
When civet SARS-CoV SZ3 was used as the query sequence in similarity plot and bootscan analysis, evidence for recombination events was also detected (Fig 5B). In the region between the two breakpoints at the genome positions nt 21161 and nt 27766, including the S gene, closer genetic relationship between SZ3 and WIV16 was observed. However, from position nt 27766 towards the 3’ end of its genome, a notably close genetic relationship was observed between SZ3 and Rf4092 instead. Throughout the non-structural gene, moreover, SZ3 shared a similarly high sequence identity with WIV16 and Rf4092. It indicates that civet SARS-CoV was likely to be the descendent from a recombinant of the precursors of WIV16 and Rf4092, or that the SARSr-CoVs found in this cave, like WIV16 or Rf4092, may have been the descendants of the SARS-CoV lineage.
Phylogenetic analysis
Phylogenetic trees were constructed using the nt sequences of nonstructural protein gene ORF1a and ORF1b. Unlike the high genetic diversity in the S gene, nearly all SARSr-CoVs from the bat cave we surveyed were closely clustered, and showed closer phylogenetic relationship to SARS-CoV than the majority of currently known bat SARSr-CoVs discovered from other locations, except YNLF_31C and 34C which were recently reported in greater horseshoe bats from another location in Yunnan [22] (Fig 6). The phylogeny of SARSr-CoVs in ORF1a and ORF1b appeared to be associated with their geographical distribution rather than with host species. Regardless of different host bat species, SARS-CoV and SARSr-CoVs detected in bats from southwestern China (Yunnan, Guizhou and Guangxi province) formed one clade, in which SARSr-CoV strains showing closer relationship to SARS-CoV were all from Yunnan. SARSr-CoVs detected in southeastern, central and northern provinces, such as Hong Kong, Hubei and Shaanxi, formed the other clade which was phylogenetically distant to human and civet SARS-CoVs (Fig 6 and S6 Fig).
Fig 6. Phylogenetic trees based on nucleotide sequences of ORF1a (A) and ORF1b (B). The trees were constructed by the maximum likelihood method using the LG model with bootstrap values determined by 1000 replicates. Only bootstraps > 50% are shown. The scale bars represent 0.03 (A) and 0.02 (B) substitutions per nucleotide position. Rs, Rhinolophus sinicus; Rf, Rhinolophus ferremequinum; Rm, Rhinolophus macrotis; Ra, Rhinolophus affinis; Rp, Rhinolophus pusillus; As, Aselliscus stoliczkanus; Cp, Chaerephon plicata. SARSr-CoVs detected in bats from the single cave surveyed in this study are in bold. Sequences detected in southwestern China are indicated in red.
Rescue of bat SARSr-CoVs and virus infectivity experiments
In the current study, we successfully cultured an additional novel SARSr-CoV Rs4874 from a single fecal sample using an optimized protocol and Vero E6 cells [17]. Its S protein shared 99.9% aa sequence identity with that of previously isolated WIV16 and it was identical to WIV16 in RBD. Using the reverse genetics technique we previously developed for WIV1 [23], we constructed a group of infectious bacterial artificial chromosome (BAC) clones with the backbone of WIV1 and variants of S genes from 8 different bat SARSr-CoVs. Only the infectious clones for Rs4231 and Rs7327 led to cytopathic effects in Vero E6 cells after transfection (S7 Fig). The other six strains with deletions in the RBD region, Rf4075, Rs4081, Rs4085, Rs4235, As6526 and Rp3 (S1 Fig) failed to be rescued, as no cytopathic effects was observed and viral replication cannot be detected by immunofluorescence assay in Vero E6 cells (S7 Fig). In contrast, when Vero E6 cells were respectively infected with the two successfully rescued chimeric SARSr-CoVs, WIV1-Rs4231S and WIV1-Rs7327S, and the newly isolated Rs4874, efficient virus replication was detected in all infections (Fig 7). To assess whether the three novel SARSr-CoVs can use human ACE2 as a cellular entry receptor, we conducted virus infectivity studies using HeLa cells with or without the expression of human ACE2. All viruses replicated efficiently in the human ACE2-expressing cells. The results were further confirmed by quantification of viral RNA using real-time RT-PCR (Fig 8).
Fig 7. Infection of Vero E6 cells by bat SARSr-CoV WIV1, Rs4874, WIV1-Rs4231S and WIV1-Rs7327S.
(A) The successful infection was confirmed by immunofluorescent antibody staining using rabbit antibody against the SARSr-CoV Rp3 nucleocapsid protein. The columns (from left to right) show staining of nuclei (blue), virus replication (red), and both nuclei and virus replication (merged double-stain images). (B) The growth curves in Vero E6 cells with a MOI of 1.0 and 0.01.
Fig 8. Analysis of receptor usage by immunofluorescence assay (A) and real-time PCR (B). Virus infectivity of Rs4874, WIV1-Rs4231S and WIV1-Rs7327S was determined in HeLa cells with and without the expression of human ACE2. ACE2 expression was detected with goat anti-human ACE2 antibody followed by fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat IgG. Virus replication was detected with rabbit antibody against the SARSr-CoV Rp3 nucleocapsid protein followed by cyanine 3 (Cy3)-conjugated mouse anti-rabbit IgG. Nuclei were stained with DAPI (49,6-diamidino-2-phenylindole).The columns (from left to right) show staining of nuclei (blue), ACE2 expression (green), virus replication (red) and the merged triple-stained images, respectively.
Activation of activating transcription factor 6 (ATF6) by the ORF8 proteins of different bat SARSr-CoVs
The induction of the ATF6-dependent transcription by the ORF8s of SARS-CoV and bat SARSr-CoVs were investigated using a luciferase reporter, 5×ATF6-GL3. In HeLa cells transiently transfected with the expression plasmids of the ORF8s of bat SARSr-CoV Rf1, Rf4092 and WIV1, the relative luciferase activities of the 5×ATF6-GL3 reporter was enhanced by 5.56 to 9.26 folds compared with cells transfected with the pCAGGS empty vector, while it was increased by 4.42 fold by the SARS-CoV GZ02 ORF8. As a control, the treatment with tunicamyxin (TM) stimulated the transcription by about 11 folds (Fig 9A). The results suggests that various ORF8 proteins of bat SARSr-CoVs can activate ATF6, and those of some strains have a stronger effect than the SARS-CoV ORF8.
Fig 9. Functional characterization of diverse ORF8 and ORF8a proteins of bat SARSr-CoVs.
(A) The ORF8 proteins of SARS-CoV and bat SARSr-CoVs induces the ATF6-dependent transcriptional activity. HeLa cells were transiently transfected with the pcAGGS expression plasmids of the ORF8 of SARS-CoV GZ02, bat SARSr-CoV Rf1, WIV1 and Rf4092 and the reporter plasmid 5×ATF6-GL3 for 40h. Control cells were co-transfected with the reporter plasmid and the empty pCAGGS vector for 24h, and treated with or without TM (2μg/ml) for an additional 16h. The cell lysates were harvested for dual luciferase assay and data are shown as the average values from triplicate wells. (B) The ORF8a proteins of SARS-CoV and bat SARSr-CoV triggered apoptosis. 293T cells were transfected with the expression plasmids of the ORF8a of SARS-CoV Tor2 and bat SARSr-CoV Rs4084 and a pcAGGS vector control for 24h. Apoptosis was analyzed by flow cytometry after annexin V staining and the percentage of apoptotic cells were calculated. Data are shown as the average values from triplicate cells. Error bars indicate SDs. * P<0.05.
Induction of apoptosis by the ORF8a of the newly identified bat SARSr-CoV
We conducted transient transfection to examine whether the ORF8a of SARSr-CoV Rs4084 triggered apoptosis. As shown in Fig 9B, 11.76% and 9.40% of the 293T cells transfected with the SARSr-CoV Rs4084-ORF8a and SARS-CoV Tor2-ORF8a expression plasmid underwent apoptosis, respectively. In contrast, transfection with the empty vector resulted in apoptosis in only 2.79% of the cells. The results indicate that Rs4084 ORF8a has an apoptosis induction activity similar to that of SARS-CoV [28].
Discussion
Genetically diverse SARSr-CoVs have been detected in various horseshoe bat species across a wide geographic range in China in the past decade [9–12,14,29]. However, most bat SARSr-CoVs show considerable genetic distance to SARS-CoV, particularly in the highly variable S1, ORF8 and ORF3 regions [10,25]. Recently, several novel SARSr-CoVs have been described to be more closely related to SARS-CoV, either in the S gene or in ORF8. The S proteins of RsSHC014, Rs3367, WIV1 and WIV16, which were reported in our previous studies, shared 90% to 97% aa sequence identities to those of human/civet SARS-CoVs [17,18]. Another strain from Rhinolophus affinis in Yunnan termed LYRa11 showed 90% aa sequence identity to SARS-CoV in the S gene [13]. In addition, two studies have described 4 novel SARSr-CoVs (YNLF_31C/34C and GX2013/YN2013) which possessed a full-length ORF8 with substantially higher similarity to that of SARS-CoV [22,30]. These findings provide strong genetic evidence for the bat origin of SARS-CoV with regard to the S gene or ORF8. However, all of these SARSr-CoVs were distinct from SARS-CoV in at least one other gene, suggesting that none of them was the immediate progenitor of SARS-CoV. Moreover, these SARSr-CoVs were discovered in bat populations from physically distinct locations. The site of origin of the true progenitor of SARS-CoV and the evolutionary origin of SARS-CoV have until now remained elusive. In the current study, we have identified a bat habitat potentially important for SARSr-CoV evolution where a series of recombination events have likely occurred among different SARSr-CoV strains, which provides new insights into the origin of SARS-CoV.
SARS first emerged in Guangdong province in late 2002 [7]. However, SARSr-CoVs discovered in bats from neighboring areas of Guangdong to date have shown phylogenetic disparity from SARS-CoV especially in the S gene [9,10,14], suggesting SARS-CoV may have originated from another region. Our analysis of the phylogeny of SARS-CoVs and all known bat SARSr-CoVs using the nt sequence of their non-structural ORF1a and ORF1b genes, which constitute the majority of the genome, shows that SARSr-CoV evolution is strongly correlated with their geographical origin, but not host species. It is noteworthy that SARSr-CoVs detected in Yunnan are more closely related to SARS-CoV than strains from other regions in China. This finding implies that Yunnan, or southwestern China, is more likely to be the geographical source of SARS-CoV than other regions in China, but data from more extensive surveillance are yet needed to support this inference.
In our longitudinal surveillance of SARSr-CoVs in a single cave in Yunnan where we discovered Rs3367, RsSHC014, WIV1 and WIV16, the CoV prevalence in fecal samples varied among different sampling time. Generally, a higher prevalence was observed in autumn (September and October) than in spring and early summer (April and May). This may be due to the establishment of a susceptible subpopulation of newborn bats which had not developed their own immunity after the parturition period [31]. Another factor may be the changes in the composition of bat species in the cave at different sampling dates. For example, in September 2012 when the CoV prevalence reached 51.3%, the majority of samples were from R. sinicus, but in May 2015 when only 3 out of the 145 samples tested positive, Aselliscus stoliczkanus was the predominant bat species in the cave. We failed to amplify the RBD sequences from 15 of the 64 SARSr-CoV positive samples. Most of these samples had comparatively low viral concentration (< 107 copies/g) (S8 Fig), as revealed by our previous quantitative studies [32]. The unsuccessful amplification of RBD in some samples with high viral concentration was probably because of the more divergent sequences in this region of these SARSr-CoV genomes.
In this cave, we have now obtained full-length genome sequences of additional 11 novel SARSr-CoVs from bats. Our findings suggest the co-circulation of different bat SARSr-CoVs highly similar to SARS-CoV in the most variable S1 (NTD and RBD), ORF8 and ORF3 regions, respectively, in this single location. In the ORF1a, ORF1b, E, M and N genes, the SARSr-CoVs circulating in this cave also shared > 98% aa sequence identities with human/civet SARS-CoVs. Thus, all of the building blocks of the SARS-CoV genome were present in SARSr-CoVs from this single location in Yunnan during our sampling period. Furthermore, strains closely related to different representative bat SARSr-CoVs from other provinces (e.g. Rs672, HKU3 and Rf1) in the RBD region were also detected there. Therefore, this cave could be regarded as a rich gene pool of bat SARSr-CoVs, wherein concurrent circulation of a high diversity of SARSr-CoV strains has led to an unusually diverse assemblage of SARSr-CoVs.
During our 5-year surveillance in this single cave, we first reported Rs3367 and WIV1 in 2013, with RBD sequence closely resembling that of SARS-CoV [17]. More recently, we discovered WIV16 which had an RBD almost identical to WIV1’s but shared much higher similarity with SARS-CoV than WIV1 in the NTD region of S1, making it the closest SARSr-CoV to the epidemic strains identified to date [18]. In this study, we found a novel strain Rs4231 from the same location sharing almost identical NTD sequence with WIV16 but distinct from it in the RBD, with evidence of a recombination event. Our recombination analysis indicated that a recombination event may have taken place at the junction between the coding region of NTD and RBD in the Rs4231 and WIV1 genomes and resulted in WIV16. Recombination at this genomic position also happened among other SARSr-CoVs relatively distant to SARS-CoV found in this location (e.g. Rs4081 and Rs4247, S5 Fig). The frequent recombination at this hotspot in the S gene increased the genetic diversity of SARSr-CoVs harbored in these bat populations and might have been responsible for the generation of the S gene of the direct progenitor strain of SARS-CoV.
The genomes of SARS-CoVs from patients during the early epidemic phase and civet SARS-CoVs all contained a single full-length ORF8 [3,7]. We have found that a number of bat SARSr-CoVs from this cave possessed a complete ORF8 highly similar to that of early human/civet SARS-CoV (>97% nt sequence identity), represented by strain Rf4092 (S3C Fig). This provided further evidence for the source of human SARS-CoV ORF8 in bats [22,30]. In contrast, the ORF8 was split into overlapping ORF8a and ORF8b in most human SARS-CoV strains from later-phase patients due to the acquisition of a 29-nt deletion [8,26]. In this study, we have discovered for the first time a bat SARSr-CoV with ORF8a and ORF8b highly similar to the later-phase human SARS-CoVs, though the split of ORF8 in the bat SARSr-CoV and that in human SARS-CoV were two independent events. Our recombination analysis suggests that this strain, Rs4084, likely acquired its ORF8 from Rf4092 through recombination, followed by the development of the 5-nt deletion which led to the splitting. It suggests that ORF8 region in bat SARSr-CoV genomes is prone to deletions as in human SARS-CoV [3,25]. Finally, the recombination analysis suggests that an ancestral strain of SARS-CoV SZ3 would have been generated if the recombination around ORF8 had occurred between the lineages that led to WIV16 and Rf4092. Taken together, the evidence of recombination events among SARSr-CoVs harbored by bats in this single location suggests that the direct progenitor of SARS-CoV may have originated as a result of a series of recombination within the S gene and around ORF8. This could have been followed by the spillover from bats to civets and people either in the region, or during movement of infected animals through the wildlife trade. However, given the paucity of data on animal trade prior to the SARS outbreak, the likely high geographical sampling bias in bat surveillance for SARSr-CoVs in southern China, and the possibility that other caves harbor similar bat species assemblages and a rich diversity of SARSr-CoVs, a definite conclusion about the geographical origin of SARS-CoV cannot be drawn at this point.
R. sinicus are regarded as the primary natural host of SARS-CoV, as all SARSr-CoVs highly homologous to SARS-CoV in the S gene were predominantly found in this species. However, it is noted that two SARSr-CoVs previously reported from R. ferrumequinum showed the closest phylogenetic position to SARS-CoV in the ORF1a/1b trees. These strains were discovered in another location in Yunnan 80 km from the cave surveyed in the current study [22]. This information also supports the speculation that SARS-CoV may have originated from this region. Nonetheless, since the correlation between the host species and the phylogeny of SARSr-CoV ORF1ab seems limited, more SARSr-CoV sequences need to be obtained from different Rhinolophus bat species in both locations in Yunnan, and from other locations in southern China. In particular, it will be important to assess whether R. ferrumequinum played a more important role in the evolution of SARS-CoV ORF1ab.
The cave we studied is located approximately 60 km from the city of Kunming. Beside a number of rhinolophid and hipposiderid species from which SARSr-CoVs have been detected, other bats like myotis were also present there. The temperature in the cave is around 22–25°C and the humidity around 85%-90%. The physical nature of the cave is not unique, but it does appear to host a particularly dense population of bats in the reproductive season. Similar caves co-inhabited by bat populations of different species are not rare in other areas in Yunnan. We propose that efforts to study the ecology, host species diversity, and viral strain populations of these caves may provide critical information on what drives SARSr-CoV evolution.
Our previous studies demonstrated the capacity of both WIV1 and WIV16 to use ACE2 orthologs for cell entry and to efficiently replicate in human cells [17,18]. In this study, we confirmed the use of human ACE2 as receptor of two novel SARSr-CoVs by using chimeric viruses with the WIV1 backbone replaced with the S gene of the newly identified SARSr-CoVs. Rs7327’s S protein varied from that of WIV1 and WIV16 at three aa residues in the receptor-binding motif, including one contact residue (aa 484) with human ACE2. This difference did not seem to affect its entry and replication efficiency in human ACE2-expressing cells. A previous study using the SARS-CoV infectious clone showed that the RsSHC014 S protein could efficiently utilize human ACE2 [33], despite being distinct from SARS-CoV and WIV1 in the RBD (S1 Fig). We examined the infectivity of Rs4231, which shared similar RBD sequence with RsSHC014 but had a distinct NTD sequence, and found the chimeric virus WIV1-Rs4231S also readily replicated in HeLa cells expressing human ACE2 molecule. The novel live SARSr-CoV we isolated in the current study (Rs4874) has an S gene almost identical to that of WIV16. As expected, it is also capable of utilizing human ACE2. These results indicate that diverse variants of SARSr-CoV S protein without deletions in their RBD are able to use human ACE2. In contrast, our previous study revealed that the S protein of a R. sinicus SARSr-CoV with deletions (Rp3) failed to use human, civet and bat ACE2 for cell entry [34]. In this study, in addition to Rs4231 and Rs7327, we also constructed infectious clones with the S gene of Rs4081, Rf4075, Rs4085, Rs4235 and As6526, which all contained the deletions in their RBD. These 7 strains, plus Rs4874 and the previously studied WIV1 and RsSHC014, could represent all types of S variants of SARSr-CoVs in this location (S3A Fig). However, none of the strains with deletions in the RBD could be rescued from Vero E6 cells. Therefore, the two distinct clades of SARSr-CoV S gene may represent the usage of different receptors in their bat hosts.
The full-length ORF8 protein of SARS-CoV is a luminal endoplasmic reticulum (ER) membrane-associated protein that induces the activation of ATF6, an ER stress-regulated transcription factor that activates the transcription of ER chaperones involved in protein folding [35]. We amplified the ORF8 genes of Rf1, Rf4092 and WIV1, which represent three different genotypes of bat SARSr-CoV ORF8 (S3C Fig), and constructed the expression plasmids. All of the three ORF8 proteins transiently expressed in HeLa cells can stimulate the ATF6-dependent transcription. Among them, the WIV1 ORF8, which is highly divergent from the SARS-CoV ORF8, exhibited the strongest activation. The results indicate that the variants of bat SARSr-CoV ORF8 proteins may play a role in modulating ER stress by activating the ATF6 pathway. In addition, the ORF8a protein of SARS-CoV from the later phase has been demonstrated to induce apoptosis [28]. In this study, we have found that the ORF8a protein of the newly identified SARSr-CoV Rs4084, which contained an 8-aa insertion compared with the SARS-CoV ORF8a, significantly triggered apoptosis in 293T cells as well.
Compared with the 154-aa ORF3b of SARS-CoV, the ORF3b proteins of all previously identified bat SARSr-CoVs were smaller in size due to the early translation termination. However, for the first time, we discovered an ORF3b without the C-terminal truncation in a bat SARSr-CoV, Rs7327, which differed from the ORF 3b of SARS-CoV GZ02 strain at only one aa residue. The SARS-CoV ORF3b antagonizes interferon function by modulating the activity of IFN regulatory factor 3 (IRF3) [27]. As previous studies suggested, the nuclear localization signal-containing C-terminal may not be required for the IFN antagonist activity of ORF3b [36]. Our previous studies also demonstrated that the ORF3b protein of a bat SARSr-CoV, termed Rm1, which was C-terminally truncated to 56 aa and shared 62% aa sequence identity with SARS-CoV, still displayed the IFN antagonist activity [37]. It is very interesting to investigate in further studies whether Rs7327’s ORF3b and other versions of truncated ORF3b such as WIV1 and WIV16 also show IFN antagonism profiles.
As a whole, our findings from a 5-year longitudinal study conclusively demonstrate that all building blocks of the pandemic SARS-CoV genome are present in bat SARSr-CoVs from a single location in Yunnan. The data show that frequent recombination events have happened among those SARSr-CoVs in the same cave. While we cannot rule out the possibility that similar gene pools of SARSr-CoVs exist elsewhere, we have provided sufficient evidence to conclude that SARS-CoV most likely originated from horseshoe bats via recombination events among existing SARSr-CoVs. In addition, we have also revealed that various SARSr-CoVs capable of using human ACE2 are still circulating among bats in this region. Thus, the risk of spillover into people and emergence of a disease similar to SARS is possible. This is particularly important given that the nearest village to the bat cave we surveyed is only 1.1 km away, which indicates a potential risk of exposure to bats for the local residents. Thus, we propose that monitoring of SARSr-CoV evolution at this and other sites should continue, as well as examination of human behavioral risk for infection and serological surveys of people, to determine if spillover is already occurring at these sites and to design intervention strategies to avoid future disease emergence.